
Die europäische Medical Device Regulation MDR (EU-Medizinprodukteverordnung) müssen Hersteller beachten, die Medizinprodukte in der EU in den Verkehr bringen wollen.
Diese Verordnung (EU) 2017/745 über Medizinprodukte, so der offizielle Titel, stellt auch Anforderungen an Benannte Stellen, Händler, Importeure und Gesundheitseinrichtungen wie Krankenhäuser.
… für Anfänger
Falls Sie ganz neu sind, dann laden Sie sich das kostenlose Starter-Kit herunter. Es verschafft Ihnen einen Überblick über die Regularien, zeigt Ihnen die Schritte zur „Zulassung“ Ihres Medizinprodukts und enthält die MDR-Checkliste als PDF und im DOCX-Format zum Download.
… für Fortgeschrittene
Steigen Sie mit dieser Seite und den darin verlinkten Fachartikeln tiefer in die Details ein.
Nutzen Sie die konsolidierte Version der MDR in Deutsch bzw. in Englisch. Diese fasst alle Änderungen an der MDR zusammen, auch die im März 2023 verlängerten Übergangsfristen. Interne Links erleichtern Ihnen die Navigation in der über 170 Seiten langen Verordnung.
… für IVDR-Hersteller
Hersteller von In-Vitro-Diagnostika sollten den Fachartikel zur IVDR lesen.
… für Importeure, Händler und Betreiber
Schauen Sie sich das folgende Video an und lesen Sie dann die in Kapitel 1.b) verlinkten Fachartikel.

Mit dem Laden des Videos akzeptieren Sie die Datenschutzerklärung von YouTube.
Mehr erfahren
1. Überblick über die Anforderungen der MDR
Dieser Artikel verwendet die Abkürzung MDR synonym zu Medical Device Regulation, EU-Medizinprodukteverordnung und „Regulation (EU) 2017/745„.
1.1 Anforderungen an Hersteller und deren Medizinprodukte
Die Anforderungen, die die Medical Device Regulation an die Hersteller richtet, sind umfangreich.
1.1.1 Produktübergreifende Anforderungen
| Anforderung der MDR und Links zu Fachartikeln | Erläuterung |
| Qualitätsmanagementsystem | Alle Hersteller benötigen ein QM-System, u. a. für die Entwicklung, Produktion und Überwachung der Produkte im Markt. Mit Ausnahme von Produkten der Klasse I ist sogar meist eine Zertifizierung notwendig. |
| Risikomanagementsystem | Das Risikomanagement muss über den ganzen Produktlebenszyklus sicherstellen, dass der Nutzen angesichts der Risiken akzeptabel ist. |
| Verantwortliche Person („Person responsible for Regulatory Compliance”) | Die Hersteller sind verpflichtet, eine Person zu beschäftigen, die für die regulatorische Konformität verantwortlich ist und z. B. sicherstellt, dass die Technische Dokumentation konform mit dem eigenen QM-System erstellt wird. |
1.1.2 Produktspezifische Anforderungen
| Anforderung der MDR und Links zu Fachartikeln | Erläuterung |
| Klassifizierung der Produkte | Für jedes Produkt muss der Hersteller die Risikoklasse festlegen. |
| Grundlegende Sicherheits- und Leistungsanforderungen und Technische Dokumentation | Die Technische Dokumentation muss den Anforderungen des Anhangs II genügen und alle Nachweise erbringen, dass die grundlegenden Anforderungen des Anhangs I erfüllt sind. Für diese Nachweise sind die Hersteller gehalten, harmonisierte Normen und die gemeinsamen Spezifikationen (Common Specifications) anzuwenden. |
| Klinische Bewertung | Im Rahmen der klinischen Bewertung muss der Hersteller kontinuierlich prüfen, ob Sicherheit, Leistungsfähigkeit und Nutzen der Produkte gegeben sind. Reichen die klinischen Daten dazu nicht aus, ist eine klinische Prüfung notwendig. |
| Unique Device Identification (UDI) | Alle Medizinprodukte müssen eine eindeutige Identifizierung erhalten, die UDI. Damit müssen die Produkte in der EUDAMED registriert werden. |
| Labeling | Die MDR legt Anforderungen an die Gebrauchsanweisung, weitere Begleitmaterialien und das sonstige Labeling wie Aufdrucke und Verpackungen genau fest. |
| Konformitätsbewertung | Abhängig von der Risikoklasse des Produkts kann der Hersteller ein Konformitätsbewertungsverfahren durchlaufen. Dabei muss er – außer bei Produkten der Klasse I – eine Benannte Stelle einbeziehen. Im Erfolgsfall muss er eine Konformitätserklärung ausfüllen und das CE-Kennzeichen anbringen. |
| Post-Market Surveillance und Vigilanz | Hersteller sind verpflichtet, ihre Medizinprodukte über deren komplette Lebensdauer im Markt zu überwachen, kontinuierlich Daten darüber zu sammeln und ggf. darauf zu reagieren. Bestehen Risiken, müssen sie die Behörden informieren. |
1.2 Anforderungen an andere Akteure (Händler, Importeure etc.)
Nicht nur die Medizinproduktehersteller müssen die EU-Medizinproduktverordnung einhalten, sondern auch andere Akteure:
- Anforderungen an Händler
- Anforderungen Importeure
- Hinweise für Krankenhäuser und andere Betreiber
- Anforderungen an Hersteller gewisser Produkte ohne medizinische Zweckbestimmung (Anhang XVI-Produkte)
Dieser Fachartikel zu den Unterschieden zwischen MDR und MDD hilft insbesondere Herstellern von Legacy-Produkten.
Wichtig besonders für diese Hersteller ist auch dieser Artikel zu den Übergangsfristen.
2. Aufbau und Struktur der MDR
Die Medical Device Regulation (EU 2017/745) umfasst 123 Artikel, die sich in 10 Kapitel gliedern. Zudem hat sie 17 Anhänge.
2.1 Die Kapitel
Die Medical Device Regulation (MDR) ist im Vergleich zur alten Medical Device Directive (MDD) völlig neu strukturiert:
| Kapitelnummer Mit Links auf MDR | Titel / Inhalt / Anforderungen Mit Links zu Fachartikeln |
| I | Anwendungsbereich und Definitionen |
| II | Wichtigste Anforderungen an Hersteller, Händler, Importeure und Mitgliedsstaaten. Viele Verweise auf Artikel in anderen Kapiteln und auf die Anhänge |
| III | Nachverfolgbarkeit von Produkten, v. a. Anforderungen zur UDI |
| IV | Anforderungen an die Benannten Stellen |
| V | Klassifizierung und Konformitätsbewertungsverfahren |
| VI | Klinische Bewertungen und klinische Prüfungen |
| VII | Post-Market Surveillance, Marktüberwachung, Meldewesen |
| VIII | Zusammenarbeit von Mitgliedsstaaten, MDCG und anderen Experten |
| IX | Vertraulichkeit, Datenschutz, Strafen |
| X | Übergangsfristen und mehr |
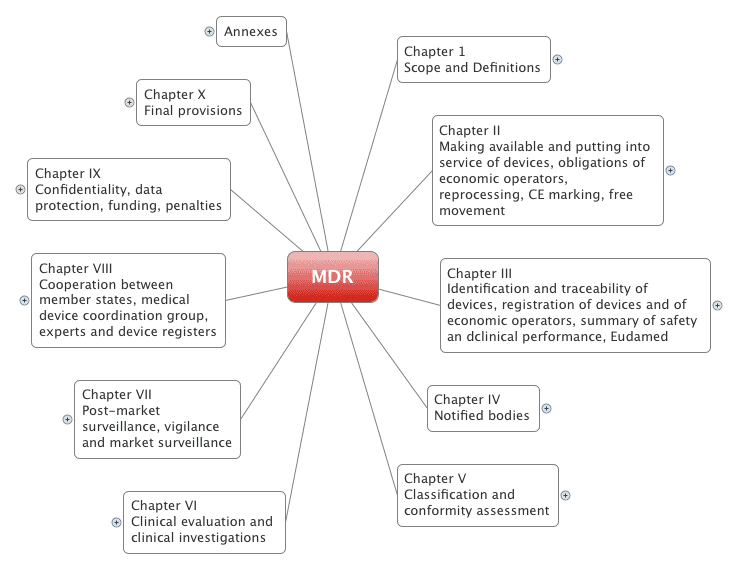
Es verschafft einen schnellen Überblick über die mehr als 170 Seiten umfassende MDR.
2.2 Die Anhänge
Die EU-Medizinprodukteverordnung hat 17 Anhänge.
| Nummer des Anhangs Mit Links zur MDR | Titel / Inhalt / Anforderungen Mit Links zu weiterführenden Informationen |
| I | Grundlegende Sicherheits- und Leistungsanforderungen |
| II | Technische Dokumentation |
| III | Technische Dokumentation für die Post-Market Surveillance |
| IV | EU-Konformitätserklärung |
| V | CE-Kennzeichnung |
| VI | UDI, EUDAMED |
| VII | Anforderungen an die Benannten Stellen |
| VIII | Klassifizierungsregeln |
| IX bis XI | Konformitätsbewertungsverfahren: – Vollständiges QM-System – Baumusterprüfung – Produktkonformitätsprüfung |
| XII | Zertifikate durch Benannte Stellen |
| XIII | Sonderanfertigungen |
| XIV | Klinische Bewertung |
| XV | Klinische Prüfung |
| XVI | Produkte ohne medizinische Zweckbestimmung |
| XVII | Entsprechungstabelle |
3. Weitere Fachartikel und Links
3.1 Weitere Fachartikel
Die EU hat die Übergangsfristen für den Umstieg auf die MDR mehrfach verschoben. Die Regeln sind so komplex, dass ein Fachartikel zu den Übergangsfristen helfen wird.
Weiter Erklärungen und Anforderungen hat die Medical Device Coordination Group (MDCG) publiziert.
Für Produkte ohne medizinische Zweckbestimmung („Anhang-XVI-Produkte“) gibt es abweichende Anforderungen.
3.2 Weitere Links
- EU 2027/745 (Originaltext)
- Deutsch (HTML) verlinkte Version des Johner Instituts (konsolidiert)
- Deutsch (HTML) Originalversion der EU
- Deutsch (PDF) Originalversion der EU
- Englisch (HTML) verlinkte Version des Johner Instituts (konsolidiert)
- Englisch (HTML) Originalversion der EU
- Englisch (PDF) Originalversion der EU
- Corrigenda
- 1. Corrigendum: VERORDNUNG (EU) 2020/561 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 23. April 2020 (bzw. auf Englisch REGULATION (EU) 2020/561 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 23 April 2020)
- 2. Corrigendum: DELEGIERTE VERORDNUNG (EU) 2023/502 DER KOMMISSION vom 1. Dezember 2022 (bzw. auf Englisch COMMISSION DELEGATED REGULATION (EU) 2023/502 of 1 December 2022)
- 3. Corrigendum: VERORDNUNG (EU) 2023/607 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 15. März 2023 (bzw. auf Englisch REGULATION (EU) 2023/607 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 15 March 2023)
- 4. Corrigendum: VERORDNUNG (EU) 2024/1860 DES EUROPÄISCHEN PARLAMENTS UND DES RATES vom 13. Juni 2024 (bzw. auf Englisch REGULATION (EU) 2024/1860 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 13 June 2024)
- Sonstiges
4. Hintergrundinformationen zur EU-Medizinprodukteverordnung
4.1 Wie es zu den EU-Verordnungen kam
Vielfach wird kolportiert, dass der Brustimplantate-Skandal Auslöser für die Überarbeitung des Medizinprodukterechts gewesen sei. Doch das bestreiten inzwischen die meisten Akteure. Damit ist weitgehend unklar, wer die Neuregulierung aus welchem Grund initiiert hat.
4.2 Unterschied von EU-Verordnungen und EU-Richtlinien
Die „alten“ EU-Medizinprodukte-Richtlinien mussten wie alle EU-Richtlinien in nationale Gesetze und nationale Verordnungen überführt werden, um Gesetzeskraft zu erlangen. Das waren in Deutschland das Medizinproduktegesetz (MPG) und Verordnungen wie die Medizinprodukte-Betreiberverordnung (weiterhin gültig) und die Medizinprodukte-Sicherheitsplanverordnung (inzwischen ungültig).
Die EU-Verordnungen (hier: MDR, IVDR) haben direkt gesetzlichen Charakter. Nationale Gesetze ergänzen diese Verordnungen nur noch, wie beispielsweise in Deutschland das MPDG u. a., um Strafvorschriften und die Festlegung der zuständigen Behörden.
4.3 Kritik an der EU-Medizinprodukteverordnung
Die Kritik an den EU-Medizinprodukteverordnungen, der Medical Device Regulation, ist massiv. Aufwände und Kosten haben sich für die Hersteller vervielfacht. Die Zeitdauern für die „Zulassungsverfahren“ ebenfalls.
Als Folge sind die Wettbewerbsfähigkeit, die Innovation und die Versorgung mit Medizinprodukten behindert.
Diese Folgen waren bereits 2020 befürchtet worden.
Mit dem Laden des Videos akzeptieren Sie die Datenschutzerklärung von YouTube.
Mehr erfahren
Für eine künftige Regulierung ist es wichtig, das System zu verstehen – eine Aufgabe der Regulatory Science.
5. Fazit und Zusammenfassung
Die Medical Device Regulation MDR, die EU Medizinprodukteverordnung, ist ein sehr umfangreiches Gesetzeswerk, das alle Akteure (Medizinproduktehersteller, Benannte Stellen, Importeure, Händler, Krankenhäuser) vor große Herausforderungen stellt. Es ist nicht erkennbar, dass die MDR die Sicherheit, Leistungsfähigkeit und Wirksamkeit der Medizinprodukte verbessert. Es ist aber erkennbar, dass die Versorgung mit Medizinprodukten leidet.
Hersteller haben kaum eine andere Wahl, als sich mit diesem Gesetzeswerk intensiv auseinanderzusetzen und dessen Anforderungen zu erfüllen. Diese erhöhten Anforderungen beschränken sich nicht auf die „Zulassung“ (Pre-market-Phase), sondern betreffen explizit auch die Post-Market-Phase (Post-Market Surveillance, Vigilanz).
Hilfe bei der Umsetzung der MDR
Kostenfreie Angebote
Haben Sie noch Fragen zur EU-Medizinprodukteverordnung MDR und zu ihrer Umsetzung? Antworten erhalten Sie in unserem kostenlosen Micro-Consulting.
Laden Sie sich das kostenlose Starter-Kit herunter, das Ihnen einen Überblick über die regulatorische Landschaft verschafft und die MDR-Checkliste als PDF und im DOCX-Format enthält.
Videos und E-Learning
Die Videotrainings im Auditgarant zeigen Ihnen Schritt für Schritt, wie Sie Ihre Technische Dokumentation und Ihr QM-System schlank, schnell und MDR-konform erstellen. Dazu stehen über 100 Templates und Musterdokumente zum Download bereit. So schaffen Sie die Voraussetzungen, um Ihre Produkte schnell und sicher zuzulassen und in den Markt zu bringen.
Produktprüfung
Die Experten des Johner Instituts helfen Ihnen beim Prüfen Ihrer Produkte:
- Usability-Tests
- Penetrations-Tests
- Test der elektrischen Sicherheit und EMV
- Prüfung der Biokompatibilität
- Klinische Bewertungen und Prüfungen
Beratung
Nutzen Sie die Expertise der Regulatory Affairs Experts des Johner Instituts, um Ihre
- regulatorische Strategie zu bestimmen und Produkte zu klassifizieren,
- Technische Dokumentation (TD) zu erstellen,
- QM-Systeme (QMS) aufzubauen,
- TD und QMS zu prüfen und Sie auf Audits und Reviews vorzubereiten sowie in diesen zu begleiten, und die
- Post-Market Surveillance zu übernehmen.
Nehmen Sie gleich Kontakt auf, damit wir gemeinsam klären können, wie Sie die regulatorischen Anforderungen der MDR schnell und einfach erfüllen und Ihre Produkte sicher in den Markt bringen können.
Änderungshistorie (ab Mai 2021)
- 2025-02-04: Leeres Hinweiskästchen zu den Synonymen am Beginn des Artikels gefüllt. Die verschiedenen Synonyme im Artikel genutzt.
- 2024-08-27: Im Kapitel 3.2 die Links zu den Corrigenda korrigiert und Links hierarchisch gruppiert
- 2024-01-18: Kapitelnummerierung geändert
- 2023-04-18: Artikel komplett neu geschrieben
- 2021-10-10: Abschnitt 7.e) (Die Konsequenzen wurden nicht betrachtet und werden nicht verstanden) eingefügt
- 2021-07-26: Link zu neuem Corrigendum ergänzt
- 2021-05-24: Neuen Beitrag zur Konformitätserklärung verlinkt



Wir bräuchten Klarstellungen in der 93/42EWG, vor allem aber im MPG, in der MPBetreibV und in der IEC 60601-1. Ein Beispiel: ME-Systeme, die bei den Betreibern zusammengestellt werden. Im MPG wird nur von Kombinationen statt von Systemen gesprochen , eine Definition dessen wovon man redet fehlt im Gegensatz zu IEC 60601-1 ganz, und was dann daraus entsteht sind individuelle Auslegungsvarianten, die dann mit Dokumenten manifestiert werden.
Die Bereinigung von vielen solchen oder ähnlichen Missständen würde vermutlich mehr bringen als eine völlig neue MRD. Statt immer nur auf „alles Neu“ in der Politik zu schielen, sollte man die Bugs beseitigen, oder wie es ein „alter Hase“ einmal gesagt hat:
Fortschritt besteht darin, dass man Gutes und Bewährtes beläßt und gleichzeitig Verbesserungswürdiges ändert, nicht in dem man überreagiert und immer alles neu fordert oder alles anders machen will.
Allgemein gesprochen, was nützen neue Gesetzte und Richtlinien, wenn schon die alten nicht eingehalten oder zweifelsfrei umgesetzt werden (können)?
Was nützen immer schärfere / aufwendige Regulatorien für Gerätehersteller, wenn man z.B. sieht, wie aus Kostengründen Systeme bei Betreibern konfiguriert werden?
Die Medizinischen Richtlinen und nationalen Gesetze sollten den Fokus beim Inhalt belassen, statt zu versuchen einige wenige kriminelle Machenschaften zu lösen. Dafür gibt es das Strafrecht.
Neue Regulatorien, die mehr Aufwand bei der Dokumentation zur Folge haben und den Fokus auch nicht mehr auf den technisch / sicherheitstechnisch relevanten Inhalt legen, bringen nur Mehraufwand.
Was fehlt sind Fachläute die die technisch / medizinischen Details beurteilen und klären können, die mehr auf den Inhalt als auf das Vorhandensein und die Form von Dokumenten, Richtlinien und Gesetzen schauen, als auf das reine Vorhandensein.
Wir haben weltweit genug / zu viel, Politiker und Manager, die sich auf das zuletzt genannte fokusierien.
Aufbereitung von Medizinprodukten / Aufbereitung von Einmalprodukten
wie ist hier der eindeutige Stand – erlaubt – nicht erlaubt – wie sehen die Anforderungen aus.
Die Wiederaufbereitung ist weiterhin erlaubt, jetzt aber genauer geregelt. Siehe z.B. Artikel 15.
Ist der Hersteller neu definiert worden? Ist das PLM-OEM-Verfahren überhaupt noch möglich wie bisher?
Sehr geehrte Frau Keil, danke für die spannende und schwierige Frage. Das Konzept der OEMs kennt die MDR jedenfalls noch. Welche Änderungen es im Detail bedeutet, ist zurzeit noch schwer abzusehen. Jedenfalls müssen die Inverkehrbringer die UDI des OEM in ihren Aufzeichnungen aufbewahren.
Wenn ich das richtig verstanden habe, darf man ein Produkt (z. B. Klasse IIa), welches die Anforderungen der MDD 93/42 erfüllt, auch noch bis zu 3 Jahre nach Inkrafttreten der MDR in Europa legal vertreiben. Wir sind ein OEM-Hersteller, welcher den Benannten Stellen des OBL nicht das Zutrittsrecht (Unannounced Audits etc.) für alle Bereiche (z. B. Produktion, Forschung) gewähren möchte. Darf unser Kunde das Produkt auch noch die nächsten Jahre vermarkten (Anforderungen MDD sind erfüllt, die der MDR nicht – da ja kein Vertrag besteht, der unlimitierte Unannounced Audits der Benannten Stelle des OBL ermöglicht).
Vielen Dank für Ihre Mühe!
Sehr geehrter Herr Böttcher,
die MDR schreibt dazu:
3a. Devices which were lawfully placed on the market pursuant to Directives 90/385/EEC and 93/42/EEC prior to the date referred to in Article 97(2) may continue to be made available on the market or put into service until five years after that date.
Das Datum sind drei Jahre nach Veröffentlichung (wahrscheinlich April 2017).
Viele Grüße, Christian Johner
Hallo, welche Auswirkungen hat die MDR auf Firmen die sich mit der Reparatur von chir. Instrumenten (Klasse 1) beschäftigen?
Ist zukünftig eine Reparatur ausschließlich den Herstellern vorbehalten?
Danke
Sie dürfen auch künftig Geräte reparieren. Allerdings werden die Hersteller stärker an die Kandare genommen: Sie müssen die Risiken auch durch eine unzureichende Wartung / Reparatur genau analysieren und beherrschen.
Gibt es die Möglichkeit, direkt Inputs in das Gesetzgebungsverfahren zu geben. Wer ist aktuell Berichterstatter des EP?
Meines Erachtens gibt es in der MDR nach wie vor fehlerhafte Aussagen in Sachen Risikobewertung. das Gesetz deckt sowohl die Sicherheit für Patienten als auch für Anwender ab.
Sowohl Anhang I, Kapitel I (1), als auch (8) implizieren, dass Risiken ‚im Vergleich zu dem für .. Anwender ..ermittelnden Nutzen vertretbar sein müssen.
Eine Risiko/Nutzen-Bewertung für Anwender (klin. Personal) widerspricht den Prinzipien des Arbeitsschutzes. Die unterschiedliche Philosophie zeigt auch der Blick in die immer noch existierende Referenz auf die Maschinenrichtlinie 2006/42/EG (warum kann man die nicht abschaffen?), deren Risikoverständnis ein anderes ist. Für den Anwender auch eines Medizinproduktes sollten keine anderen Bedingungen gelten, und auch für Patienten sollte das Sicherheitsniveau unabhängig vom Nutzen sein, außer bei Risiken, die unmittelbar mit einer Anwendung zu tun haben (Röntgenstrahlung, Schneidevorgang Skalpell, u.ä.). Nur deshalb wurde von der EN1441 zur EN ISO 14971 der Teil der Risiko/Nutzen-Bewertung hinzuaddiert.
Sehr geehrter Herr Schraag,
danke für Ihre Rückmeldungen! Direkt zu Ihren Fragen:
Sehr geehrter Herr Prof. Johner,
‚Anwender‘ ist auf Seite 60 des MDR drafts definiert. Da gibt es wenig Interpretationsspielraum. In der Einleitung, beispielsweise Punkt 2 ist die Zielsetzung (Sicherheit für Patienten und Anwender) auch klar definiert. Dies entspricht auch der ursprünglichen Absicht, nur ein Gesetz (bzw. Richtlinie im Fall der MDD) für Medizinprodukte zu haben, mit dem Konformitätsausweis also alle Sicherheitsaspekte abzudecken. Dies gilt auch für die IEC 60601-1, die im Ammendment 3.1 zwar noch zur Definition ‚Patient‘ erläuternd hinzugefügt hat, dass dieser auch ‚Operator‘ sein kann. Aber mit der Erfüllung der 60601-1 gilt auch die Sicherheit der Anwender als gewährleistet. Selbstverständlich sind die Grenzwerte (bsp. Leckströme) und Sicherheitsmaßnahmen an die besondere Situation der Patienten angepasst (man muss auch die Hilflosigkeit berücksichtigen, angefangen vom Neugeborenen bis zum anästhesierten Patienten).
Die Erfüllung einer harmonisierten Norm entspricht dem allgemein anerkannten Stand der Technik, gilt als akzeptables Risiko ohne irgendwelche Abwägung des Nutzens. Diese Situation ist adäquat der sowohl eines beliebigen Haushaltsgeräts, als auch einer Maschine im Sinne der Maschinenrichtlinie, bei denen es ebenfalls keine Abwägungen gegen den Nutzen gibt (Alle ISO/IEC Normen folgen dem Guide 51, ursprünglich auch die ISO 14971).
Das Besondere bei Medizingeräten ist, dass für den Zweck von Behandlung, Diagnose, usw. Einflussnahmen auf den menschlichen Körper notwendig sind, deren Risiken zwar minimiert werden können, aber auch Verletzungen zur Folge haben (können). In diesen Fällen müssen Arzt / Patient die Notwendige Information haben, um informierte Entscheidungen treffen zu können, müssen klinische Daten existieren, die zeigen, dass ein Nutzen höher sein kann als das Risiko (ähnlich Medikamenten).
Dies trifft aber nicht für den Anwender zu (schließen wir mal den Sonderfall Anwender = Patient aus). Die MDR impliziert, dass auch für den Anwender eine Nutzen-Abwägung getroffen wird, eben weil sie nicht differenziert, wie Sie das versuchen. Das geht aber nicht, es widerspricht unseren ethischen Grundwerten.
Eine Bemerkung zur Maschinenrichtlinie: Diese ist eher ein Unfall im Gesetzgebungsverfahren 2007, durchgesetzt in letzter Minute, weil eine Abgeordnete des EP von einem Todesfall im Zusammenhang mit einer fehlerhaften Anwendung von Luer-Konnektoren erfahren hatte. Das historisch gewachsene Problem der Luer-Konnektoren kann leider nicht durch die Maschinenrichtlinie gelöst werden, ist aber seit vielen Jahren in intensiver Arbeit durch das entsprechende ISO-Normierungskommitee. Ansonsten lassen sich alle mechanischen Gefahren durch die MDD/MDR abdecken, ohne Ausflug zur Maschinenrichtlinie.
Das alles hilft dem Hersteller natürlich wenig, er muss mit dem leben,was ihm Kommission und Parlament vorsetzen. Siehe auch die content deviations der EN ISO 14971:2012. Nur wäre es besser, wenn man rechtzeitig solche Mängel adressieren könnte, als sich im Nachhinein aufwendig darum herum winden zu müssen..
Hallo Herr Johner,
übernimmt die qualifizierte Person die Aufgaben und Verpflichtungen des früheren Safety Officers for Medical Devices? Wie sieht es hier speziell in Fragen der (persönlichen) Haftung der qualifizierten Person aus? Gibt es ggf. Unterschiede, je nachdem an welchem Standort die qualifizierte Person in Europa (von Interesse sind hier auch die Sonderrollen Schweiz und bald UK) ansässig ist?
Vielen Dank für Ihre Interpretation.
Sehr geehrter Herr Sattler,
ja, davon gehe ich aus: Die qualifizierte Person und der Sicherheitsbeauftragte werden personenidentisch sein.
Die persönliche Haftung einer angestellten(!) qualifizierten Person lebt immer bei grob fahrlässiger oder vorsätzlicher Handlung wieder auf. Sonst bleibt es die des Arbeitgebers.
Die Haftbarkeitsfragen werden in unterschiedlichen Ländern unterschiedlich bewertet und v.a. auch mit Strafen bewehrt. Das ist immer Sache der Nationalstaaten.
Beim Standort ist das Arbeitsverhältnis zu klären. Ist es ein entsandter Mitarbeiter, Ex Pat oder lokal angestellter Mitarbeiter.
Das UK und Schweizer-Recht bezüglich Strafen kennen ich nicht gut genug, um profund antworten zu können.
Beste Grüße, Christian Johner
Wie sieht die neue MDR ein „Software Standalone“-Produkt angewandt im Health-care Bereich.? Konkret, wie werden LIS (Laboratory Information System, auch LIMS genannt) eingestuft werden?
Momentan ist die Lage nicht klar (Klasse I, nicht steril, ohne Messeigenschaft), die Interpretationen sind unterschiedlich.
Geht die MDR auch auf die Frage ein, was geschieht, wenn diese SW als APP anwendbar ist oder ist dies widerrum eine andere Thematik (E-Health Gesetzgebung)
Vielen Dank
Petra Pilgram
An der Begriffsdefinition ändert die MDR im Vergleich zur MDD nichts. Wenn Ihr LIS „nur“ der Dokumentation dient, ist es kein MP, wenn es der Diagnose dient, dann fällt es darunter.
Falls es ein MP wäre, würde sich die Klasse wahrscheinlich ändern (von I auf IIa oder höher).
Die MDR unterscheidet dabei nicht, ob die Software eine App ist. Falls es eine App wäre, verlangt sie aber, dass man die Plattform spezifiziert und die spezifischen Risiken betrachtet.
Die E-Health-Gesetzgebung hat mit den medizinprodukterechtlichen Anforderungen nichts zu tun. Leider gibt es ein paar dubiose Anbieter, die in gesetzeswidriger Weise den Eindruck erwecken, es gäbe spezielle Zertifikate.
Ich hoffe, ein wenig geholfen zu haben. Falls nicht, schreiben Sie mir einfach.
Christian Johner
Sehr geehrter Herr Johner,
etwas Verwirrung gibt es, da ich unterschiedliche Aussagen zu der Übergangsfrist gehört habe. Die einen sagen, alle Geräte müssen nach den drei Jahren erneut zugelassen werden, andere sagen, es dürfen vorher (vor Inkrafttreten der MDR) zugelassene Geräte weiterverkauft werden. Wie ist es denn nun richtig? So wie ich Sie verstehe und die MDR es vermuten lässt, müssen nur Geräte, die ab Inkrafttreten der MDR zugelassen/markteingeführt werden, die Anforderungen der MDR erfüllen. Ist dies so richtig?
Viele Grüße,
Sebastian Barrenstein
Sehr geehrter Herr Barrenstein, danke für die sehr wichtige Frage.
Ich plane demnächst einen Blogbeitrag dazu. Damit Sie nicht solange warten müssen, schicke ich Ihnen gleich per Mail eine Antwort.
Beste Grüße, Christian Johner
Welche Verpflichtungen oder Möglichkeiten ergeben sich für einen Gesundheitsdiensteanbieter wie beispielsweise ein Krankenhaus durch die Kennzeichnungspflichten (z.B. Chargendokumentation, Abfrage der Produkt- und Herstellereigenschaften, etc.
Die UDI trifft erst einmal die Hersteller.
Manufacturers shall comply with the obligations relating to the UDI system referred to in Article 27 and with the registration obligations referred to in Articles 29 and 31.
Dabei ist ein Hersteller:
‚manufacturer‘ means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark;
Als Eigenhersteller müssen Sie allerdings für Folgendes sorgen:
(ii) the details necessary to identify the devices;
Das muss aber kein UDI sein.
Für Geräte, die Sie einkaufen, müssen Sie die UDI-Anforderungen nicht erfüllen. Die MDR schreibt zum Thema Logistikkette nur:
(c) any health institution or healthcare professional to which they have directly supplied a device.
Das richtet sich aber an die Hersteller. Für die Krankenhäuser gilt:
Health institutions shall store and keep preferably by electronic means the UDI of the devices which they have supplied or with which they have been supplied, if those devices belong to class III implantable devices.
For devices other than class III implantable devices, Member States shall encourage, and may require, health institutions to store and keep, preferably by electronic means, the UDI of the devices with which they have been supplied.
Member States shall encourage, and may require, healthcare professionals to store and keep preferably by electronic means, the UDI of the devices with which they have been supplied with.
Sehr geehrter Herr Prof, Johner,
in Artikel 10 (14) steht „Die Hersteller händigen der zuständigen Behörde auf deren Ersuchen hin alle Informationen und Unterlagen, die für den Nachweis der Konformität des Produkts erforderlich sind, in einer von dem betreffenden Mitgliedstaat festgelegten Amtssprache der Union aus.“
Normalerweise hätte ich ‚zuständige Behörde‘ als diejenige des Landes interpretiert, in der der Hersteller niedergelassen ist und sich registriert hat.
Artikel 10 (14) und auch andere Stellen unterscheiden aber klar: „Die zuständige Behörde des Mitgliedstaats, in dem der Hersteller seine eingetragene Niederlassung hat, kann verlangen, dass …“
Im schlimmsten Fall kann man dies so lesen, dass jede zuständige Stelle innerhalb der EU Unterlagen einfordern kann, und dass diese übersetzt werden müssten. Unterlagen, die den Konformitätsnachweis erbringen können sehr umfangreich sein, inkl. der Testberichte von Testhäusern.
Wissen Sie, ob die Differenzierung im Text bewusst und so gewollt ist? Wenn ja, dann wäre das ein schwer kalkulierbares Geschäftsrisiko.
Mit freundlichem Gruß,
Martin Schraag
Danke für Ihre Frage, lieber Herr Schraag!
Damit wird auch auf Firmen Bezug genommen, die außerhalb der EU sitzen. Ein Hersteller würde sich nur in einem Land registrieren.
Dass den Behörden mehr Rechte eingeräumt werden, sehe ich allerdings schon.
Die MDR ist sicher kein Freunde der kleinen Firmen…
Viele Grüße, Christian Johner
Ja, Herr Prof. Johner,
für kleine Firmen könnte die MDR das Aus bedeuten, auch wenn es ein paar wenige Erleichterungen gibt (Bsp. ‚verantwortliche Person‘).
Zu Ihrer letzten Antwort: Der Bezug auf Firmen außerhalb der EU ist in Artikeln 11 bis 13 geregelt, wobei in Artikel 11 d), e) ebenfalls zwischen zuständiger Behörde allgemein und der im Land der Niederlassung unterschieden wird.
Ich hatte meine Anfrage parallel an das ZLG gestellt, habe bisher aber keine klärende Auskunft erhalten. Ich kann mir keinen Reim darauf machen, außer es gab im PIP-Fall die Situation, dass Anfragen von Behörden außerhalb Frankreich nur mit Dokumentation in Französisch beantwortet wurden, und man dem nun vorbeugen will. Praktisch mag das zwar selten ein Problem sein, wenn die Behörden zwischen den EU-Ländern effektiv kommunizieren. Wenn es aber zuschlägt muss der Hersteller im schlimmsten Fall erst mal ein Übersetzungsbüro für ein paar Monate buchen.
Sehr geehrter Herr Prof. Johner,
ich habe inzwischen Antwort von der ZLG bekommen. Vorbehaltlich einer dortigen Rückfrage mit direkt an den Verhandlungen Beteiligten wird meiner obigen Interpretation zugestimmt: Der Hersteller muss in Zukunft auf Aufforderung Unterlagen an beliebige nationale Behörden in deren Amtssprache liefern. ‚Unterlagen‘ kann alles sein, was zum Konformitätsnachweis gehört.
Es wurde in diesem Zusammenhang auch auf Artikel 93 und 99 verwiesen.
Guten Tag Herr Prof. Johner,
Mit welchen Auswirkungen haben Contract Manufacturer zu rechnen? Die Verantwortung liegt ja dabei beim Inverkehrbringer des Produktes, jedoch werden bestimmt auch Anforderungen an die Lieferanten gestellt?
Vielen Dank für Ihren ausführlichen Bericht und die sehr interessanten Antworten in der Kommentarliste.
freundliche Grüsse
Raoul BRem
Sehr geehrter Herr Brehm,
danke für Ihre positive Rückmeldung!
Die Anforderungen an „Contract Manufacturers“ ergeben sich, wie Sie schreiben, indirekt aus den Verpflichtungen der Inverkehrbringer. Für die ist nicht nur die MDR künftig zu beachten, sondern noch früher und mindestens so speziell die Anforderungen der ISO 13485:2016. Hier sind Qualitätssicherungsvereinbarungen schriftlich zu vereinbaren, deren Inhalte aus dem Risikomanagement abgeleitet sind. Als „Contract Manufacturer“ stellen Sie einen ausgelagerten Prozess dar.
Viele Grüße
Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
unsere Bank hält einen Defibrillator vor (im Kundenbereich). Welche Auswirkungen hat die neue MDR für uns als Bank? Welche Maßnahmen müssen wir ergreifen um der MDR gerecht zu werden?
Freundliche Grüße
Eckhard Zingel
Sehr geehrter Herr Zingel,
die kurze Antwort wäre: Keine zusätzlichen.
Sie sind keine Gesundheitseinrichtung, insofern betrifft Sie die MDR kaum. Die Anforderungen der Herstellers z.B. an Überprüfung, Wartung oder ggf. Austausch bleiben davon aber unberührt.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
können Sie sagen, ob die neue MDR für den Bereich UDI letztendlich einen (einzigen) Standard für die eindeutige Identifizierung von Medizinprodukten herbeiführen wird? Müssen sich bisherige Institutionen wie GS1 oder EHIBCC/ HIBC letztlich „einigen“?
Vielen Dank für Ihre Einschätzung!
MfG
Thorsten Riedel
Sehr geehrter Herr Riedel,
es ist unwahrscheinlich, dass man sich auf einen einzigen Standard beschränken wird, weil sich dahinter kommerziell arbeitende Firmen verbergen. Ein Monopol zu gewähren, wäre wettbewerbswidrig.
Viele Grüße, Christian Johner
Hallo Herr Johner,
wie sieht es denn aus bei schon vorhandenen Kosemtikgeräten? Darf man diese noch weiter verwenden und wie lange? (Mesotherapiegeräte, Koagulationsgeräte, Plasma Pen). M.f.G.
Sehr geehrte Frau Herbst-Weidenfeld,
Kosmetikgeräte, die bisher keine Medizinprodukte waren, fallen nur dann unter die Regularien der MDR, wenn sie in Anhang XVI genannt sind. Scrollen Sie dazu ganz ans Ende der MDR.
Beachten Sie, dass diese Liste von der EU ohne parlamentarischen Prozess geändert werden kann.
Viele Grüße
Christian Johner
Sehr geehrter Herr Johner,
die Idee mit dem Berechner der Zertifikatslaufzeit ist wirklich großartig. Ich habe unser CE-Zertifikat (bis 30.06.2018) mal eingegeben, aber es kam ein ganz anderes Ergebnis (30.06.2022) raus als ich vermutet hatte. Ich bin davon ausgegangen dass wir unser CE-Zertifikat in 2018 nochmal verlängern können (also theoretisch bis 30.06.2025) und das aufgrund der Begrenzung in der MDR die Zertifikate bis zum 27.05.2024 gültig wären.
Könnten Sie mir bitte erläutern warum der Rechner auf den 30.06.2022 kommt und ob ich die MDR falsch interpretiere?
Vielen Dank und viele Grüße
Daniela Hauff
Sehr geehrte Frau Hauff,
es gibt wahrscheinlich keinen Widerspruch: Wenn Sie Ihr Zertifikat verlängern, dann ist es das Datum des verlängerten Zertifikats, das Sie in den Rechner eingeben müssen. Das kann der Rechner nicht wissen, ob und für wie lange Sie verlängern.
Geben Sie bitte Bescheid, wenn das die Frage nicht beantwortet haben sollte. Ich bessere dann gerne nach.
Nochmals vielen Dank für die wichtige Frage!
Beste Grüße, Christian Johner
Eine weiterführende Frage zu Lims & Lis ist, ob diese Software nicht doch ein Medizinprodukt wird bzw. ist.
Jedes Lims & Lis hat berechnete Parameter, die zur Diagnose dienen und weiters hat ein Lis & Lims Qualitätskontrolle integriert.
Dies würde ein Medizinprodukt von min. Klasse II ergeben oder nicht?
Mit der MDR kann es durchaus sein, dass die LIMS bzw. LIS als Medizinprodukte klassifiziert werden, zumal es den (zugegeben auch überraschenden Ausschluss) wie in der IVD nicht gibt.
Der Trend bei den LIM- bzw. LI-Systemen geht zudem in die Richtung, dass immer mehr diagnostische Funktionen beinhaltet sind, was eine Klassifizierung als Medizinprodukt zwangsläufig macht.
Danke für Ihren wichtigen Hinweis, lieber Herr Tomasini.
Hallo, mich würde mal interessieren wer genau die Verfasser der MDR sind.
Welche Behörden, Institutionen usw. waren beteiligt und welche Unternehmen waren eventuell beratend tätig?
Die Verfasser ist v.a. die EU-Kommission. Natürlich gab es Lobby-Arbeit (die Schlimmeres verhinderte) und Einflüsse der Normengremien, Verbände und benannten Stellen, aber das Schreiben verbleibt bei der EU und deren Juristen.
Sehr geehrtes Johner-Institut-Team,
ist die EN 62304 weiterhin harmonisiert zur MDR?
Hierzu habe ich im Amtsblatt der EU keinen Hinweis gefunden.
Oder werden die Normen erst noch angepasst, bevor diese als harmonisiert gelten?
Ich wünsche Ihnen einen sonnigen Tag und freue mich auf Ihre Antwort.
Mit freundlichen Grüßen
Marco Bransch
Sehr geehrter Herr Bransch,
derzeit ist noch keine Norm harmonisiert. Zuerst müssen die Z-Anhänge geschrieben werden, die angeben, wie vollständig die Norm die Anforderungen abdeckt.
Mit den besten Grüßen, Christian Johner
Sehr geehrter Herr Johner,
welche Auswirkungen hat die MDR auf eine ZSVA/ AEMP.
Muss ich als Aufbereiter alle Instrumente (auch Klasse I) im Haus verfolgen können?
Muss jedes Instrument (Klasse I) eine UDI besitzen?
MfG
Mark Galander
Sehr geehrter Herr Galander,
die Auswirkungen in der MDR betreffen die Hersteller bzw. Inverkehrbringer von Medizinprodukten.
Die Anforderungen der MDR an die UDI gelten unabhängig von allen Produkten. Ob Sie Produkte im Haus verfolgen könne müssen, ist so explizit nicht geregelt. Das hängt auch von den Anforderungen des Herstellers ab. Wenn der z.B. die Anzahl der Aufbereitungszyklen limitiert, müssen Sie als „Aufbereiter“ die Produkte identifizieren und ggf. aussortieren können.
Welche nationalen Vorgaben und Ergänzungen zur MDR veröffentlicht werden, insbesondere solche, die sich an die Betreiber werden, weiß ich nicht.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
wie ist der Begriff „placing on the market“/“Inverkehrbringen“ zu verstehen? Per Definition ist es „the first making available“/“die erstmalige Bereitstellung“. Was aber genau meint das?
Ist damit das allererste Produkt gemeint, das ich auf den Markt bringe, und kann ich somit nur genau 1x ein Produkt erstmalig bereitstellen?
Oder meint es das erstmalige Bereitstellen in Bezug auf die Lieferkette? Also meint es den Wirtschaftsakteur, der in der Wirtschaftskette der erste ist und somit die erstmalige Bereitstellung durchführt (daher per Definition dem Hersteller/Importeur vorbehalten)? Bezieht sich Inverkehrbringen auf jedes einzelne Produkt, das vom Band läuft und kann ich ein Produkt somit immer wieder erstmalig bereitstellen?
Oder ist damit etwas völlig anderes gemeint?
Mit dem richtigen Verständnis dieses Begriffes wird die Interpretation der Überprüfungsanforderungen an den Importeur interessant:
„In order to place a device on the market, importers shall verify that…“. Muss er es ein einziges Mal tun oder immer wieder und für jedes einzelne Produkt?
Vielen Dank im Voraus für Ihre Einschätzung.
Viele Grüße
Jennifer Ehmcke
Sehr geehrte Frau Ehmcke,
die MDR definiert es wie folgt:
„Inverkehrbringen“ bezeichnet die erstmalige Bereitstellung eines Produkts, mit Ausnahme von Prüfprodukten, auf dem Unionsmarkt;
D.h. der Importer muss nicht für jedes einzelne Produkt prüfen.
Hilft das? Falls nicht, einfach nachhaken.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof Johner,
wie verhält sich die klinische Bewertung bzw klinische Prüfung der Mdr bei Sonderanfertigungen der Risikoklasse 1? Es exestieren viele einzelne Versionen dieser Produkte, und noch mehr Produktkategorien. Klinische Daten werden ja aus klinischen Prüfungen, oder aus der Literatur vergleichbarer Produkte gezogen. Da diese Produkte meistens keine „gleichwertigen“ Produkte im Markt haben, ist die Informationslage sehr dünn. Wird von den Herstellern verlangt für zb. Orthesen für jedes Produkt eine klinische Prüfung durchzuführen? Oder werden die Hausinternen Daten, die nach dem Inverkerhbringen und im Arbeitsalltag generiert werden ausreichen?
Vielen Dank.
Sehr geehrter Herr Wetzelsperger,
Sie müssen eine klinische Bewertung Ihrer Produkte durchführen. Da gibt es keine Ausnahmen. Allerdings gibt es u.U. eine Ausnahme bei der Forderung, dass diese anhand klinischer Daten erfolgen muss. Ob das bei Orthesen der Fall wäre, weiß ich nicht, ich habe aber Zweifel.
Die Post-Market-Daten können und müssen Sie nutzen. Wenn damit ausreichende klinische Daten vorliegen, um die Sicherheit, Leistungsfähigkeit und den klinischen Nutzen zu beweisen, benötige Sie auch keine klinische Prüfungen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre Antwort. Ich muss leider noch mal nachhaken. Das heißt also, die Definition bezieht sich nicht auf die Reihenfolge des Wirtschaftsakteurs in der Lieferkette, sondern auf das allererste Mal, das der Importeur sein Produkt auf dem EU-Markt bereitstellt. Also muss er seinen Überprüfungspflichten nur ein einziges Mal nachkommen? Nämlich in dem Moment, in dem er zum ersten Mal das Produkt vom nicht EU-Hersteller erhält und bevor er es an einen Händler oder Endkunden weitergibt, richtig?. Danach nie wieder? Sicherlich aber bei Änderungen am Produkt oder nach Ablauf von Zertifikaten.
Herzlichen Dank und viele Grüße
Jennifer Ehmcke
Sehr geehrte Frau Ehmcke,
danke für Ihr Nachfragen. Wir sollten die unterschiedlichen Aufgaben des Importeurs mit dem EU-Bevollmächtigten im Blick behalten.
Sie beziehen sich auf den Artikel 12 Absatz 2 der MDR. Diese verlangt vom Importeur „nur“ ein paar grundlegende Prüfungen. Ein Importeur kann nicht für jedes einzelne Produkt überprüfen, ob es ein CE-Zeichen trägt. Denken Sie an einen Importeur, der Hunderttausende Spritzen einführt. Es ist auch sehr unwahrscheinlich, dass ein Hersteller gerade bei Massenprodukten bei einem die CE-Kennzeichnung „vergisst“.
Wenn ein Importeur Grund zur Annahme hat, dass ein Hersteller Schwierigkeiten hat, die Anforderungen zu erfüllen, muss er reagieren. Generell haftet aber primär der EU-Repräsentant.
Eine Änderung am Produkt ist ein neues Produkt und bedeutet eine neue UDI. Damit muss der Importeur die Prüfung erneuern.
Die Überprüfung von Zertifikaten und weiteren Unterlagen wie die technische Dokumentation sehe ich eher beim EU-Repräsentant.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
Wir sind Zulieferer in die Medizintechnik Branche. Wir liefern Materialien für Endprodukte nahezu aller Risikoklassen.
Dazu habe ich 2 Fragen:
1.) In wie weit betrifft die MDR Zulieferer? So weit ich das verstanden habe, betrifft Zulieferer die erhöhte Anforderung an die Dokumentation, aber welche Art der Dokumentation muss der Zulieferer zur Verfügung stellen?
2.) Sie erwähnend ei Anforderung an ein Qualitätsmanagement System gemäß ISO 13485:2016. Wie wird diese Anforderung begründet? Ich konnte in der MDR keine direkte Verpflichtung zur ISO 13485 herauslesen, d.h. würde eine Zertifizierung nach zB ISO 9001 ebenfalls die Anforderung erfüllen? Bzw. gilt dies auch für Zulieferer?
Vielen Dank im Voraus!
mfg
Kristin Kraker
Sehr geehrte Frau Kraker,
danke für Ihre Fragen!
ad 1.) Sie als Zulieferer sind nur indirekt betroffen. Wahrscheinlich werden Ihre Kunden erhöhte Anforderungen stellen z.B. an die Dokumentation, an die Möglichkeit zur Auditierung und an die Kommunikation. Welche Dokumentation Ihr Kunde verlangt, kann ich in Unkenntnis des Produkts nicht sagen. Die Anforderungen an die technische Dokumentation sind zwar deutlich umfangreicher geworden. Wer allerdings normenkonform gearbeitet hat, dürfte keine großen Änderungen vornehmen müssen.
ad 2.) Als Zulieferer gibt es keine Anforderungen an das QMS aus der MDR. Die Hersteller sind bei den meisten Konformitätsbewertungsverfahren dazu verpflichtet. Diese werden bei ausgelagerten Prozessen entweder ihr eigenes QMS auf Sie überstülpen (zumindest für die entsprechenden Prozesse) oder von Ihnen ein zertifiziertes QMS verlangen. Die ISO 13485 wird die harmonisierte Norm für QMS sein nicht die ISO 9001. Eine direkte Verpflichtung zu einem QMS für die Zulieferer nennt die MDR nicht.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
in dem Artikel 20 der MDR geht es um die CE Kennzeichnung von Medizinprodukten.
Halten Sie es für notwendig, dass wiederverwendbare chirurgische Instrumente generell zusätzlich zur CE Kennzeichnung auch mit der Nummer der benannten Stelle gekennzeichnet werden müssen. Wenn ja, gibt es hier Fristen bis wann die alte, nur „CE“ Kennzeichnung zulässig ist und ab wann zusätzlich die Nummer der benannten Stelle auf dem Instrument aufgebracht sein muss? Gäbe es hier dann „Ausnahmen“ für bereits schon mal inverkehr gebrachte Produkte?
Danke.
Herzliche Grüße
Ralph Wonner
(5) Wo erforderlich, wird der CE-Kennzeichnung die Kennnummer der für die Konformitätsbewertungsverfahren gemäß Artikel 52 zuständigen Benannten Stelle hinzugefügt.
Sehr geehrter Herr Wonner,
danke für Ihre Frage! Ich halte es für gesetzlich notwendig, dass die Anforderungen des Artikel 20 eingehalten werden, was die CE-Kennzeichnung mit einschließt, bei Klasse Ir-Produkten auch die Nummer der benannten Stelle. Sie zitieren genau den relevanten Artikel und Absatz.
Produkte, die bereits in Verkehr gebraucht wurden, dürfen gemäß der Übergangsfristen weiter bereitgestellt und Inbetriebgenommen werden. Eine weitere Nutzung ist nicht untersagt.
In der Hoffnung geholfen zu haben und mit vielen Grüßen, Christian Johner
Sehr geehrter Herr Johner,
auf der Seite:
https://www.johner-institut.de/blog/regulatory-affairs/medical-device-regulation-mdr-medizinprodukteverordnung/
unter:
Drohendes Marktverbot -> Artikel 73 sagt die MDR „elektronisches System für klinische Prüfung“
LG A.Sander
Sie haben Recht, Herr Sander!
Der Text bezog sich auf eine alte Version der MDR. Hier lautete Artikel 73 „Amtliche Feststellung der Nichtkonformität“. Diese Forderungen finden sich jetzt in den Artikel 94ff. Den Artikel muss ich noch anpassen.
Danke für den Hinweis!
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof Johner,
danke für Ihre Neuigkeiten, die immer wieterhelfen. Bin gerade aber etwas ratlos:
Im Corrigendum der MDR heißt es,
Artikel 120 Absatz 10
(10)Produkte, die gemäß Artikel 1 Absatz 6 Buchstabe g in den Geltungsbereich dieser Verordnung fallen und die nach den vor dem 26. Mai 2020 in den Mitgliedstaaten geltenden Regeln rechtmäßig in Verkehr gebracht oder in Betrieb genommen wurden, dürfen in den betreffenden Mitgliedstaaten weiterhin in Verkehr gebracht und in Betrieb genommen werden.
der Buchstabe (f) – tierisches Gewebe fällt demnach raus.
Meines Erachtens wiederspricht Absatz 4 des Artikels 120 aber dem Absatz 10:
(4)Produkte, die vor dem 26. Mai 2020 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden, und Produkte, die ab dem 26. Mai 2020 aufgrund einer Bescheinigung gemäß Absatz 2 des vorliegenden Artikels in Verkehr gebracht wurden können bis zum 27. Mai 2025 weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.
Was halten Sie von diesem Widerspruch?
LG Peter Postert
Sehr geehrter Herr Postert,
Sie stellen eine großartig und schwierige Frage, auf die ich noch keine Antwort habe. Ich mache mich mal kundig und gebe Bescheid, wenn ich auf eine Lösung stoße.
Es tut mir leid, dass ich ausnahmsweise nicht (sofort) helfen kann.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
die in der oben stehenden „Downloads“-Box gelisteten verlinkten Dokumente in englischer und deutscher Sprache enthalten noch die Informationen vor Veröffentlichung des Corrigendums 15409/1/18 REV 1.
Zur einfacheren Arbeit mit der MDR möchte ich daher gerne zwei Verbesserungsvorschläge anmerken:
1. Verlinkung des Corrigendums in der oben stehenden „Downloads“-Box
2. Übernahme der Korrekturen des Corrigendums direkt in die verlinkten Dokumente
Vielen Dank und beste Grüße,
Thore Bosk
Sehr geehrter Herr Prof Johner,
wir interpretieren die Änderung des Artikel 120 Absatz 10 etwas anders:
Produkte, die gemäß Artikel 1 Absatz 6 Buchstabe g in den Geltungsbereich der MDR fallen, sind:
„…Produkte, die aus Derivaten von Geweben oder Zellen menschlichen Ursprungs hergestellt sind, die nicht lebensfähig sind oder abgetötet wurden.“
Dies sind Produkte, die unter der MDD grundsätzlich nicht als Medizinprodukte europaweit zuzulassen waren, sondern nur „nach den … in den Mitgliedstaaten geltenden Regeln“ rechtmäßig in den Verkehr gebracht und in Betrieb genommen wurden.
Diese Produkte dürfen weiterhin „nach den … in den Mitgliedstaaten geltenden Regeln“ in den Verkehr gebracht und in Betrieb genommen werden.
(Die ebenfalls in Artikel 1 Absatz 6 Buchstabe g erwähnten „Transplantate, Gewebe oder Zellen menschlichen Ursprungs und ihre Derivate, die unter die Richtlinie 2004/23/EG fallen…“ können sowieso nicht als Medizinprodukte zugelassen werden, da sie ja per Definition Arzneimittel sind.)
Nun wurden – wohl versehentlich (?) – auch „Produkte … gemäß Artikel 1 Absatz 6 Buchstabe f“ eingeschlossen:
„…Produkte, die aus Geweben oder Zellen tierischen Ursprungs oder ihren Derivaten hergestellt sind, die nicht lebensfähig sind oder abgetötet wurden“
Dies sind Produkte, die man auch schon unter der MDD grundsätzlich als Medizinprodukte europaweit zulassen konnte – und somit eine nationale Zulassung „nach den … in den Mitgliedstaaten geltenden Regeln“ zur Marktfähigkeit nicht notwendig war. Daher wurden diese Produkte im Corrigendum des Absatz 10 gestrichen.
Somit sind für letztere Produkte die Fristen Artikel 120 Absatz 2 („Bescheinigungen, die von Benannten Stellen vor dem 25. Mai 2017 gemäß den Richtlinien 90/385/EWG und 93/42/EWG ausgestellt wurden, bleiben bis zu dem in der Bescheinigung angegebenen Zeitpunkt gültig, außer im Fall von Bescheinigungen gemäß Anhang 4 der Richtlinie 90/385/EWG bzw. gemäß Anhang IV der Richtlinie 93/42/EWG, die spätestens am 27. Mai 2022 ihre Gültigkeit verlieren“) und Absatz 4 („Produkte, die vor dem 26. Mai 2020 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden, und Produkte, die ab dem 26. Mai 2020 aufgrund einer Bescheinigung gemäß Absatz 2 des vorliegenden Artikels in Verkehr gebracht wurden können bis zum 27. Mai 2025 weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.“) maßgeblich.
VG, Christoph Piechaczek
Sehr geehrter Herr Professor Johner,
vielen Dank an dieser Stelle für die hervoragende Aufarbeitung der Themen zur MDR.
Anbei eine Frage: Inwieweit gilt die Dokumentations- und Umsetzungspflicht mit UDI etc.für ein Krankenhaus bei Kleinimplantate, wie Schrauben, die aus einem Schrauben-Sieb stammen .
In so einem Sieb sind gut und gerne an die 100 kleine Schrauben enthalten. Wie sollen diese zukünftig dokumentiert werden? Eine UDI für jede einzelne Schraube? Gibt es dazu auch Vorgaben?
Vielen Dank und beste Grüße
K. Winkels
Sehr geehrter Herr, sehr geehrte Frau Winkels,
die UDI-Anforderungen richten sich primär an die Hersteller, nicht an die Gesundheitseinrichtungen, auch wenn der Gesetzgeber die durchgängige Nachverfolgbarkeit von Artikeln wünscht. Da Sie vermutlich nicht der Hersteller dieser Schrauben sind, müssen Sie keine UDI-Vorgaben erfüllen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Antwort, heißt dies im Umkehrschluss, dass die Art und Außmaß der Dokumentationspflicht des Krankenhauses von Bereitstellung einer UDI pro Einzelschraube abhängt? Sofern der Hersteller keine eindeutige Kennung für die Einzelschraube bereitstellt, kann das Krankenhaus wie gehabt patientenbezogen dokumentieren?
Beste Grüße
Katja Winkels
Sehr geehrte Frau Winkels,
so ist es! Und selbst wenn der Hersteller eine eindeutige Kennung anbringt, folgen aus der MDR (noch) keine konkreten Anforderungen an Sie.
Sie erfüllen übrigens bereits den eigentlichen Gedanken der MDR, wenn Sie patientenbezogen dokumentieren. Damit ist die „Traceability“ des Produkts von der Herstellung bis zu dessen Verwendung gegeben.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner, ich möchte gerne die Frage von Frau Winkes vom 7. Juni19 08:53 und Ihre Antwort vom 11.06.19 18:48 vertiefen.
… „Inwieweit gilt die Dokumentations- und Umsetzungspflicht mit UDI etc. für ein Krankenhaus bei Kleinimplantate, wie Schrauben, die aus einem Schrauben-Sieb stammen.In so einem Sieb sind gut und gerne an die 100 kleine Schrauben enthalten.“. Schlüpfe ich als Aufbereiter in der Rolle des Herstellers wenn ich diese hunderte von Schrauben immer wieder aufs neu reinige, desinfiziere, sterilisiere? Zweite Frage: gibt es eine Regelung im MDR, die dieses ständiges PORBLEM für die Aufbereiter endlich klar stellt? Aus nationalem Recht haben wir als Aufbereiter sehr wohl die Pflicht lückenlos zu dokumentieren, was in dem Beispiel unmöglich ist:( UDI hin und her.
Vorab vielen herzlichen Dank und Grüße,
D. Eissing
Sehr geehrter Herr Prof. Johner,
genau Ihre Aussage kann ich leider nicht begreifen:
„Sie erfüllen übrigens bereits den eigentlichen Gedanken der MDR, wenn Sie patientenbezogen dokumentieren. Damit ist die „Traceability“ des Produkts von der Herstellung bis zu dessen Verwendung gegeben.“
MDR schreibt von Aufbereitung, Neuaufbereitung, Weiterverwendung von MP.
Fragen: 1. Was ist mit der „Wiederaufbereitung? Wenn ich hunderte von Schrauben in einem Sieb habe ein paar Jahre immer wieder Neu?- wieder?- aufbereite.
2. Wie Lang ist das Lebenszyklus dieser Produkte?
3. Das hat dann nichts mehr mit „von der Herstellung bis zu dessen Verwendung“ zu tun, oder denke ich falsch?
Vielen Dank nochmals,
Herzliche Grüße, D. Eissing
Sehr geehrte Frau Eisenring,
danke für Ihr Nachhaken. Ich bin nicht ganz sicher, ob ich jede Ihrer Fragen richtig verstehe. Dennoch wage ich eine erste Antwort:
Die MDR definiert einen Hersteller als
Alles das tun sie als Krankenhaus nicht. Sie machen keine „Neuaufbereitung“, sondern eine „Aufbereitung“. Eine Aufbereitung ist laut MDR
Nach meinem Verständnis reinigen Sie, desinfizieren und sterilisieren Sie Schrauben. Das ist eine Aufbereitung und keine Herstellung, zumal sie die Schrauben nicht „unter ihrem eigenen Namen oder ihrer eigenen Marke vermarkten“. Sie sind also kein Hersteller. Die Anforderungen der MDR, auch die bezüglich UDI, richten sich primär an Hersteller, nicht an Betreiber.
Ich widerspreche nicht, dass es weitere nationale Anforderungen an die Betreiber gibt, auch die Sterilisation betreffend.
Die Lebensdauer der Schrauben legt der Hersteller fest. Diese startet mit einer Sterilisation oder Reinigung nicht von neuem. Ideal wäre es, wenn der Hersteller nicht nur die Lebensdauer, sondern auch die Anzahl der Aufbereitungszyklen festlegt.
Falls ich Ihre Fragen noch nicht ausreichend präzise beantwortet haben, dann nutzen Sie gerne unser Micro-Consulting, das Sie auf unserer Homepage finden. Bitte beziehen Sie sich dann auf dieser Konversation.
Beste Grüße, Christian Johner
Guten Morgen Herr Prof. Johner,
zu der Aussage:
„Sie erfüllen übrigens bereits den eigentlichen Gedanken der MDR, wenn Sie patientenbezogen dokumentieren. Damit ist die „Traceability“ des Produkts von der Herstellung bis zu dessen Verwendung gegeben“ hätte ich ein paar Fragen.
1. Wie kann ich patientenbezogen bei hunderte von Schrauben und Platten in einem Sieb, die jahrelang immer wieder gereinigt, desinfiziert und sterilisiert werden, dokumentieren.
2. Wie kann ich die o.g. Produkte nachvollziehbar in der Aufbereitung rückverfolgen?
3. Wenn ich eine Schraube heute auspacke, in einem Sieb hineinlege und diese zwei Jahre lang immer wieder aufs neu aufbereite, dann begreife ich nicht wie ist das mit der Rückverfolgbarkeit/ Lebenszyklus „von der Herstellung bis zu dessen Verwendung“?
4. MDR und die nationalen Gesetze und Verordnungen sprechen von Aufbereitung, Neuaufbereitung, Weiterverwendung, Einmalprodukte… Unter welchem Begriff und Artikel MDR fällt eigentlich die Wiederaufbereitung von Einmalprodukte? Oder denke ich falsch?
Vorab vielen Dank für Ihre Antwort.
Herzliche, D. Eissing
…vielen Dank für Ihre Antwort Herr Prof. Johman.
Gute Woche für Sie, D. Eissing
Sehr geehrter Herr Prof. Johner,
vielen Dank für den MDR-Übergangsfristen-Rechner. Wie sieht es denn in den Fällen aus, in denen Produkte aus Klasse I höher – in Klasse IIa oder IIb – eingestuft werden müssen, da es sich um stoffliche Produkte handelt? Da gibt es bisher ja keine Bescheinigung einer Benannten Stelle. Dürfen diese Produkte nach dem 27.05.2020 nur dann weiterhin bereitgestellt (= weiter verkauft) werden, wenn sie bis zu diesem Termin eine Bescheinigung einer Benannten Stelle erhalten haben?
Oder trifft auch auf diese Produkte der folgenden Passus zu?
„Produkte, die vor dem 26. Mai 2020 gemäß den Richtlinien 90/385/EWG und 93/42/EWG rechtmäßig in Verkehr gebracht wurden ….können bis zum 27. Mai 2025 weiter auf dem Markt bereitgestellt oder in Betrieb genommen werden.“
Vielen Dank schon vorab.
Mit freundlichen Grüßen, U. Rosche
Sehr geehrte Frau Rosche,
es ist wie Sie vermuten: Wenn Sie keine Bescheinigung haben, und das Produkt durch die MDR höher gestuft wird als Klasse I, dann dürfen Sie das Produkt nicht mehr in den Verkehr bringen.
Der genannte Passus hilft nichts, weil es um die Bereitstellung und Inbetriebnahme geht. Die Bereitstellung ist nicht der Verkauf. Am besten Sie Studieren unsere Seite zur Inverkehrbringung. Die Sache ist nicht ganz banal.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
wir stellen Medizinprodukte der Klasse IIa her. Die Zertifikate der benannten Stelle für diese Produkte werden verlänger. Somit haben wir eine Übergangszeit bis Mai 2024.
Trotzdem müssen wir die MDR-Anforderungen hinsichtlich Vigilanz, PMS, PMCF, klinischer Bewertung und der verantworlichen Person bereits ab Mai 2020 erfüllen.
Ist das richtig? Gibt es noch weitere Anforderungen der MDR, die ab Mai 2020 gefordert werden?
Vielen Dank für Ihre Hilfe!
Mit freundlichen Grüßen, K.H. Suddera
Sehr geehrter Herr Suddera,
Ihr Einschätzung teile ich!
Was man noch in Betracht ziehen könnte, ist die Thematik UDI / EUDAMED, weil das wiederum für die Vigilanz wichtig sein kann. Je nach Konstellation sollten Sie die Anforderungen an die Wirtschaftsakteure (Importeur, Distributor, EU-Repräsentant) beachten.
Mein Haupt-Tipp wäre, sicher zu stellen, dass die Anforderungen der MDD(!) tatsächlich sauber erfüllt sind. Ich sage das, weil wir bei den meisten unserer „MDR-Transitionsprojekte“ v.a. erst einmal die MDD-Compliance erreichen müssen. In anderen Worten: Momentan werden viele Altlasten auf die Kostenstelle MDR gebucht.
Beste Grüße, Christian johner
Vielen Dank für die rasche Antwort!
Ich kann nur bestätigen, dass es bei uns auch einige Altlasten aufzuarbeiten gibt. Aber es ist beruhigend zu hören, dass es bei Anderen auch so ist.
Vielen Dank für die Hilfe und schöne Grüße,
K.H. Suddera
Sehr geehrter Herr Prof. Dr. Christian Johner.
Mit großem Interesse habe ich den mir alle Fragen und Antworten bezogen auf den MDR angeschaut und mir ein „Bild“ gemacht.
Meine Frage ist bezogen auf den MEDIKALHANDEL.
Also was hat eine Händler zu beachten, der direkt beim Hersteller bezieht, konsequent Chargenkontrollen, Sichtkontrollen und MHD Kontrollen durchführt.
NICHT Inverkehrbringer ist, sondern nur mit den Produkten handelt. Was hat der MDR für diese Unternehmen an Bedeutung.
Freue mich von Ihnen zu lesen.
Mit freundlichem Gruß
Stefan Möller
Sehr geehrter Herr Möller,
die MDR stellt im Artikel 14 die Anforderungen an die Händler.
Weitere Pflichten wie die zur Meldung können sich aus Verträgen mit den Herstellern ergeben. Weiter gibt es Anforderungen an Medizinprodukteberater, die nicht die MDR, sondern da MPG bzw. das künftige MDG formulieren.
Viele Grüße, Christian Johner
Guten Abend Prof. Johner,
als Anwender von MP stell ich mir die Frage, ob ich ein gültig erworbenes MP ( z.B: eine steril verpackte Bipolarelektrode), nach dem CE Ablaufdatum noch verwenden darf.
(Bsp. CE ausgestellt am 01.01.2016/ heute wäre der 02.02.2021)
Der Artikel 120 MDR, beschreibt dies für mich recht diffus.
Ebenso die Frage 18 aus dem Fragekatalog der NAKI
https://www.bundesgesundheitsministerium.de/fileadmin/Dateien/3_Downloads/N/NAKI/NAKI_02-05_UG1_MDR_FAQ_UEbergang.pdf
Sehr geehrter Herr Wiest,
die Übergangsfristen der MDR wenden sich wie die MDR selbst an die Hersteller und andere sogenannte Wirtschaftsakteure, nicht aber an Anwender.
Ein Hersteller insbesondere von steilen Produkten sollte das Ablaufdatum festlegen. Solange können Sie das Produkt nutzen. Das gilt auch, wenn dieses Datum nach dem 25. Mai 2020 liegt. Die relevante Frist ist die für die Inbetriebnahme: Die ist definiert als:
Diese Frist ist der 27.05.2025.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
wir sind Hersteller von Medizinprodukten der Klasse I, also Inverkehrbringer. Unsere Kunden sind Fachhändler (Sanitätshäuser), die wiederum Inbetriebnehmen, indem sie das Produkt dem Endkunden bereitstellen. Die Inbetriebnahme bzw. Bereitstellung auf dem Markt, von Produkten nach MDD, ist bis zum 27.05.2025 möglich.
Oft ist es so, dass ein Medizinprodukt vom Endkunden zurück an den Fachhandel geht, dort hygienisch aufbereitet wird und erneut in Betrieb genommen wird. Das Produkt kommt zu einem neuen Endanwender. Ist dies nach dem 27.05.2025 nicht mehr möglich? In Artikel 120, Absatz 4 steht leider nicht „erstmals“ in Betrieb genommen.
Vielen Dank vorab!
Mit freundlichen Grüßen,
Maren Bromm
Sehr geehrte Frau Bromm,
Sie haben Recht, die MDR spricht nicht von „erstmaligen Inbetriebnehmen“. Dieses Konzept kennt die Verordnung nicht – nur das des erstmaligen Inverkehrbringens. Solange es sich um eine Aufbereitung eines Produkts handelt, wird es nicht neu in den Verkehr gebracht. D.h. Sie hätten keine Thematik mit den Übergangsfristen. Es wäre nur sicher zu stellen, dass es keine „Neuaufbereitung“ ist.
Beste Grüße, Christian Johner
Hallo Hr. Dr. Johner,
eine kurze Frage bzgl. des letzten Woche veröffentlichten Corrigendums. Auch nach mehrmaligen Durchlesen des Art. 120 (3) und (4) und Auseinanderpflücken des Textes ist uns immer noch nicht endgültig klar, welche Produkte der Klasse I davon betroffen sind.
So wie wir das jetzt lesen, trifft dies für Produkte zu, die derzeit Klasse I sind und zukünftig höher klassifiziert werden (z. B. Medical Software Applications).
Es trifft damit nicht für Produkte zu, die unter der MDD Klasse I sind und auch zukünftig in der Klasse I unter der MDR bleiben. Wenn dem so wäre, müßte für die absolut risikoärmsten Medizinprodukte die MDR zum Mai 2020 umgesetzt sein. Ist diese Sichtweise korrekt?
Vielen Dank vorab für eine kurze Kommentierung!
Herzliche Grüße
Wolfgang Decker
Sehr geehrter Herr Decker,
ich teile genau Ihre Sichtweise: Die MDR gewährt Klasse-I-Produkten, die künftig in höhere Klassen (z.B. Klasse Ir) fallen die Übergangsfrist. Das betrifft auch Software. Allerdings ändern Hersteller ihre Software so häufig, dass der „Bestandsschutz“ schnell fallen wird.
Pflichten, wie die nach Post-Market-Surveillance konform MDR, gelten für alle Produkte ohne Übergangsfrist.
Sehr geehrter Herr Prof.Johner,
können Sie absehen, welche Pflichten auf Apotheken zukommen? Vielen Dank & freundliche Grüße,
Lars Frohn
Sehr geehrter Herr Frohn,
danke für die spannende Frage!
Welche Pflichten für Apotheken gelten, hängt von deren Tätigkeit ab. Ich sehe v.a. die Händlerpflichten als die relevanten. Der Artikel 14 ist hier besonders wichtig.
Weiter werden künftig auch Pflichten im Kontext der UDI auf die Apotheken zukommen.
Beste Grüße, Christian Johner
Guten Morgen Herr Johner, wie ist diese Anpassung des Art. 5 zu interpretieren?
(b) verify that the certificate for the single-use device has not been suspended, withdrawn or subject to restrictions (gestrichen- from the market) as indicated in the European Database on Medical Devices (EUDAMED) when the relevant system will be available, oras well as on the website of the manufacturer or of its authorised representative
(c) verify if the use of the single-use device has been subject to(gestrichen- that the single-use device is not subject to restrictions for safety) reasons as indicated in the field safety notices available on EUDAMED, or, in case EUDAMED is not fully functional on 26 May 2020, on the system(s) or procedures that will be relied upon for the purpose of meeting the relevant obligations regarding exchange of information laid down in Regulation (EU) 2017/745, until the obligations and requirements that relate to EUDAMED will apply in accordance with Article 34 and 123(3) of Regulation (EU) 2017/745, (gestrichen-EUDAMED) or as well as on the website of the manufacturer or its authorised representative;
Herzlichen Dank für Ihre Rückmeldung, Herr Eissing
Sehr geehrte Herr Eissing,
ich brauche Ihre Hilfe, um Antworten zu können. Sie beziehen sich auf welchen Absatz im Artikel 5?
Ich sehe bei Artikel 5 nur einen Abschnitt „c“ in dem es heißt
Ich sitze wahrscheinlich auf den Augen :-).
Beste Grüße, Christian Johner
Guten Morgen Herr Prof. Johner,
gibt es schon die erste Ergebnisse zum Thema: Common specifications für die Aufbereitung Einmalartikeln erarbeiten geplant für Q4 2019?
Danke und Grüße, Mariane Maier
Sehr geehrte Frau Maier,
danke für Ihre Frage. Ich habe bisher von keinen konkreten Ergebnissen erfahren. Sobald ich etwas weiß, werde ich es im Instituts-Journal melden.
Ich grüße Sie herzlich, Christian Johner
Guten Morgen Herr Prof. Johner,
dass die Übergangsfristen den Hersteller betreffen, ist bei mir schon längst angekommen. Trotzdem habe ich die Verständnisfrage an Sie:
In wie fern/ Zusammengang soll mich das Thema: Übergangsfristen, als Klinik- Betreiber/ Anwender oder Aufbereiter (Reinigung, Desinfektion, Sterilisation) von MP beschäftigen?
Vielen Dank für Ihre Klarstellung.
Beste Grüße, Anton Maier-Hofer
Sehr geehrter Herr Maier-Hofer,
die Anforderungen an die Aufbereitung von Einmalprodukten regelt der Artikel 17. Für den sieht die MDR keine Übergangsfristen vor. Sie erlaubt jedoch für Gesundheitseinrichtungen besondere Festlegungen durch die Mitgliedsländer. Das ist im Fall von Deutschland das MPDG. Dieses sieht aber auch keine Übergangsfristen dafür vor. D.h. dass beispielsweise eine Anzeigepflicht besteht:
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
wir sind Lohnhersteller von Medizinprodukten der Klassen 1 und 2a. Die Produkte werden
a) im Auftrag entwickelt, die komplette technische Dokumentation an die Kunden übergeben. Alleine diese kümmern sich um die Anmeldung als auch sämtliche Formalismen deren Produkte. Unsere Kunden sind alleinige Inverkehrbringer. Wir treten nach außen nicht in Erscheinung.
b) für die Kunden lediglich abgefüllt (Bereitstellung der Bulkware durch den Kunden). Ansonsten wie unter a) beschrieben
c) für die Kunden hergestellt (Fullservice), jedoch unter Produktionsvorgaben durch den Kunden. Ansonsten wie unter a) beschrieben.
Meine Frage lautet jetzt dahingehend, ob sich nun auf Grund der MDR (eine OEM-PLM-Konstellation soll ja mit in Kraft treten der MDR nicht mehr möglich sein) für uns etwas an der bisherigen Konstellation ändert (z.B. Nennung des Originalherstellers auf der Packung)
Für eine erschöpfende Antwort wäre ich Ihnen sehr dankbar.
Mit freundlichen Grüßen,
Karl Kohlert
Sehr geehrter Herr Kohlert,
ich sehe keine Änderung für Lohnhersteller.
Ich vermute, dass Sie unter dem QM-Dach des Inverkehrbringers arbeiten bzw. über Qualitätssicherungsvereinbarungen von dem an die Kandare genommen sind. Sie sind – so verstehe ich Ihre Frage – nicht der Legalhersteller. Somit wenden sich die Pflichten der MDR nicht an Sie. Die Anforderungen der MDR gelten v.a. für Hersteller, Händler, Importeure und Bevollmächtigte sowie Gesundheitseinrichtungen.
Dass die Hersteller gewissen Anforderungen an Sie übertragen, steht auf einem anderen Blatt. Rechnen Sie auch verstärkt mit Lieferantenaudits.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ich habe eine Frage zu „offenen“ Geräten, wie z.B. PCR-Automaten oder ELISA-Geräten, mit denen verschiedene Testkits verschiedener Hersteller kombiniert werden können. Gibt es laut IVDR eine Anforderungen, ob und wie sichergestellt werden soll, dass Gerät in Kombination mit Testkit valide Ergebnisse liefert? Sollten das die Hersteller in den Gebrauchsanweisungen angeben?
Liebe Frau Hend,
ja, die IVDR fordert in den grundlegenden Sicherheits- und Leistungsanforderungen (Anhang I Kap II) folgendes:
„13.1. Wenn ein Produkt zur Verwendung in Kombination mit anderen Produkten oder Ausrüstungen bestimmt ist, muss die Kombination einschließlich der Verbindungen sicher sein und darf die vorgesehene Leistung der Produkte nicht beeinträchtigen. Jede Einschränkung der Anwendung im Zusammenhang mit solchen Kombinationen wird auf der Kennzeichnung und/oder in der Gebrauchsanweisung angegeben.“
Wir IVD-Experten empfehlen, die in der Leistungsbewertung Ihres Produktes kreuz-validierten Kombinationen anzugeben und künftig ggf. erweiterte validierte Kombinationen über einen permanenten Link auf Ihrer Webseite zu ergänzen. Die Bewertung und Kontrolle der Risiken, die durch die verschiedenen Kombinationen einhergehen, sollten im Risikomanagement dokumentiert werden. Beste Grüße Sebastian Grömminger
PS: Vielen Dank für die Möglichkeit, hier Fragen stellen zu dürfen!
Mit freundlichen Grüßen
Ingrid Hend
Sehr geehrter Herr Prof. Johner,
Betreffend der Klassifizierung von Software: Laut MPG ist das Inverkehrbringen von nicht registrierten/nicht CE markierten Medizinprodukten strafbar. Wie sieht das aus bei falsch klassifizierten Medizinprodukten, wie zum Beispiel einer Software, welche als MDR Klasse I selbstzertifiziert wird, die Behörden bei einer Inspektion jedoch der Meinung wären, es könnte eine Klasse IIa oder höher sein?
Herzlichen Dank
Eva
Sehr geehrte Eva,
es ist auch ein Gesetzesverstoß, ein Produkt falsch zu klassifizieren, weil man damit nicht dar richtige Konformitätsbewertungsverfahren durchlaufen hat.
Zudem kann das auch als Verstoße gegen das Gesetz gegen den unlauteren Wettbewerb geahndet werden.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner
Im Anhang II 3b.) heisst es, „vollständige Informationen und Spezifikationen einschließlich der Herstellungsprozesse und ihrer Validierung…“
Anhang II 3c) sagt „Angabe aller Stellen, einschließlich Lieferanten und Unterauftragnehmer, bei denen Auslegungs- und Herstellungstätigkeiten durchgeführt werden.“
Kann man diese beiden Punkte zusammenbringen und rechtfertigen, dass auch alle Lieferanten, welche nur Komponenten für das MP liefert, ebenfalls eine Prozessvalidierung machen müssen? Oder sind diese beiden Punkte getrennt zu betrachten?
Vielen herzlichen Dank für Ihre Antwort.
Mit freundlichen Grüssen
Alfred M.
Sehr geehrter Herr M.
die beiden Punkte sind erst einmal getrennt zu betrachten. Dass aber kritische Lieferanten gelenkt werden müssen fordert die MDR u.a. im Anhang IX, vergleichbare Anforderungen hat die ISO 13485.
Die generelle Forderung nach einer Prozessvalidierung beim Lieferanten gibt es nicht. Gerade bei Katalogware sieht man typischerweise davon ab. Bei kritischen Bauteilen sieht das natürlich anders aus.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
muss ein Händler von Medizinprodukten die gesamte UDI auf den Lieferscheinen angeben?
Würde es zum Zweck der Nachvollziehbarkeit auch reichen, wie bisher geschehen, die Artikelnummer, die Lotnummer und das Ablaufdatum anzugeben bzw. aus der UDI herauszusuchen und anzugeben?
Wenn nein, ab wann wird die Angabe von UDIs auf Lieferscheinen verpflichtend sein?
Der Hintergrund zur Frage ist, dass wir überlegen, mit der UDI auf Scanner und entsprechende Programme umzusteigen, die die gescannte UDI automatisch in unser Warenwirtschaftsprogramm zuordnen werden.
Dafür möchte wir wissen, ob und ab wann das notwendig sein wird.
Herzlichen Dank!
Sehr geehrte Frau Hend,
die Pflicht zur Nachverfolgbarkeit der Produkte im Markt ergibt sich aus dem Artikel 25: Dort heißt es:
Damit ergibt sich keine Pflicht, die UDI aus dem Lieferschein anzugeben. Andere Möglichkeiten das Produkt nach- bzw. rückverfolgen zu können, bestehen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Hilfe!
Da dieses Thema für uns sehr wichtig ist und wir die richtigen Entscheidungen treffen möchten, sind noch weitere, präzisiere Fragen dazugekommen.
Trifft die Möglichkeit, auf dem Lieferschein keine UDI anzugeben, sondern stattdessen wie bisher Lotnummer etc., auf Produkte aller Risikoklassen zu?
Sind ausschließlich die Produkte der Klasse III, die als sogenannte „Implantate“ bezeichnet werden, elektronisch mit der UDI zu erfassen und zu speichern, oder betrifft diese Pflicht alle Klasse III Produkte?
Könnten zukünftig mit der UDI-Pflicht für Klasse II und Klasse I-Produkte auch für Händler und Importeure eine neue Verpflichtung dazu kommen, die UDI elektronisch zu erfassen und zu speichern?
In Artikel 27 (8) und (11) steht außerdem, dass Die Kommission im Wege von Durchführungsrechtsakten weitere Produkte, Produktkategorien oder Produktgruppen festlegen kann, bei denen dann sehr wohl die UDI erfasst und gespeichert werden muss.
Unter welchen Umständen könnte das passieren? Welche Produkte, Produktekategorien oder Produktgruppen könnten davon betroffen werden?
Vielen Dank für Ihre Hilfe!
Neulassungen in der Interimszeit (so sie denn kommt)
Sehr geehrter Herr Johner,
einmal mehr, Danke für ihren Blog!
Wie muss man bei der Konformitätserklärung vorgehen, findet die MDR-Verschiebung tatsächlich statt?
Wenn TDs (Klasse I, pur) noch nicht MDR-tauglich sind, doch für die Interimszeit MDD-Doc ausstellen, obwohl die MDD ja ausläuft? Muss das Produkt vom Markt? Läuft die MDD wirklich Ende Mai 2020 aus oder verlängert sich dieser Termin dann ebenfalls?
Was machen die Hersteller neuer Produkte (z. B. IIb) mit vollständiger MDR-TD, wenn sie keinen NB finden? Produkt monatelang „liegen lassen“?
Ich denke, diese Fragen wird die EU bald klären, aber man/frau denkt ja voraus …
Beste Grüße,
Cornelia Platte
Das ist eine spannende Frage!
Die Lage ist momentan so: Die Verschiebung der MDR ist empfohlen, noch nicht beschlossen aber wahrscheinlich. Das MPDG, das das MPG aufhebt, ist verabschiedet, vom Bundespräsidenten aber nicht unterzeichnet. Ich hoffe, dass das MPDG nicht in Kraft tritt, weil wir sonst einen gesetzlichen Widerspruch hätten: MPG aufgehoben, MDR nicht in Kraft.
Ich würde mich so verhalten, als würde das MPG und das MDD noch ein Jahr weiter gelten. Entsprechend würde ich die Konformität mit MDD erklären. Gleichzeitig würde ich sicherstellen, dass alles bereits MDR-konform ist. Denn es gilt ja auch „Stand er Technik“.
Wenn Sie die Benannte Stelle hängen lässt, dann melden Sie sich. Wir helfen dabei, Dinge zu eskalieren. Sie bekommen Ihr TD-Review und Ihr MDR-Audit rechtzeitig.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
sollte das MPDG im Mai vom Bundespräsidenten unterschrieben werden und somit dann offiziell das MPG ablösen, dann hätten wir (nach meinem Wissensstand) de facto keinen Sicherheitsbeauftragten mehr, da dieser von der „verantwortlichen Person“ der MDR abgelöst werden würde. Allerdings sehe ich genau da die Diskrepanz die Sie bereits erwähnt haben, da die MDR (zumindest höchstwahrscheinlich) um ein jahr verschoben wird und somit die Rechtsgrundlage fehlt. Wir haben den Vertrag unseres externen Sicherheitsbeauftragten bereits vor ein paar Monaten ab dem Zeitraum Juni 2020 gekündigt und fragen uns nun, ob wir das rückgängig machen sollten… Gemäß ihrer Aussage sollten wird genauso weitermachen wie bisher, als ob MPG und MDD noch in Kraft wären, unabhängig vom MPDG oder nicht, jedoch fehlt dann in der nationalen Rechtfassung der Sicherheitsbeauftragte… Können Sie für uns die Sache evtl. aufklären?
LG Tobias Bauer
Lieber Herr Bauer,
ich verstehe Ihre Frage, habe aber noch keine Antwort. Wir wissen erst Bescheid, wenn die MDR endgültig verschoben und das MPDG unterschrieben ist, ob wir das Problem wirklich haben. Ich vermute, dass man das MPDG nochmals anhält bzw. die Daten ändert.
Sobald ich Weiteres weiß, gebe ich im Instituts-Journal Bescheid.
Danke für Ihre spannende und sehr relevante Frage!
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
wie kann man offiziell Erfahren das die MDR verschoben wird/ist?
MfG
Jens Amberg
Sehr geehrter Herr Amberg,
ich publiziere den offiziellen Stand mit dem nächsten Instituts-Journal. Am Wochenende bereite ich das auf.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
unsere benannte Stelle hat uns einen Ergänzungsvertrag zum ZertVertrag geschickt, der die Belange der MDR ergänzt. So weit, so gut.
Nun hat sie diesen Vertrag aufgrund der MDR-Verschiebung aktualisiert, v.a. bzgl der Datumsangaben. Ein Passus blieb aber ungeändert:
„Der Auftraggeber gewährleistet, dass seine Produkte weiterhin der MDD bzw. AIMDD entsprechen und er ab 26. Mai 2020 sämtliche Anforderungen der MDR an die Überwachung nach dem Inverkehrbringen, die Marktüberwachung sowie die Vigilanz entsprechend Art. 12(3) MDR umsetzen und erfüllen wird.“
Müsste es hier nicht auch „2021“ heißen?
Mit freundlichen Grüßen
Ralf Egenolf
Korrektur zu meiner Anfrage von 15:00 Uhr:
Art. 120 (3) muss es natürlich heißen.
Mit freundlichen Grüßen
Ralf Egenolf
Sehr geehrter Herr Egenolf,
Sie haben absolut Recht: Es sind alle Fristen, die 25./26.05.2020 um ein Jahr verschoben. Daher vermute ich, dass es ein redaktioneller Fehler Ihrer BS ist.
Die anderen Übergangsfristen verschieben sich aber nicht um ein Jahr. Defacto ist dadurch die Übergangsfrist um ein Jahr kürzer.
Viele Grüße, Christian Johner
Guten Tag Herr Johner
Wir stellen unter anderem wiederverwendbare Instrumente in einer Charge her. Die Chargen Nr. ist auf dem Instrument im HRI Format aufgebracht.
Gemäss MDR muss zukünftig jedes Instrument eine einmalige Serial-Nr. aufweisen.
Frage:
Müssen nun die Instrumente welche wir ab Mai 2021 in Verkehr bringen eine Serial-Nr. aufweisen?
Wenn ja, muss diese im HRI-Format ab diesem Zeitpunkt auf dem Instrument aufgebracht sein?
Wie steht dies im Zusammenhang der Artikel 123 Buchstabe g) (Übergangsfrist für UDI-Träger DPM)?
Besten Dank für Ihre Rückmeldung und freundliche Grüsse
Peter Ackeret
Guten Tag Herr Ackeret,
vielen Dank für Ihre Frage dich in Vertretung von Professor Johner gerne beantworte.
Generell gilt die Forderung der Angabe einer Seriennummer nur für aktiv implantierbare Medizinprodukte gemäß MDR 23.1 s).
Für die Anbringung der UDI gibt es Übergangsfristen. Bei wiederverwendbaren Instrumenten der Klasse Ir muss die Direktmarkierung erst für ab dem 26. Mai 2027 in Verkehr gebrachte Produkte erfolgen (siehe Artikel 123 (3) (g)).
Herzliche Grüße
Luca Salvatore
Guten Tag Herr Johner,
sehe ich es richtig, dass Medizinproduktehersteller die ein Konformitätsbewertungsverfahren nach Anhang V-MDD durchlaufen haben sich nun nach Anhang XI-Teil A (MDR) oder theoretisch nach Anhang IX (MDR) bewerten lassen können?
Noch eine Frage zu Anhang XI-Teil A Abschnitt 10.1 – Bedeutet dies, dass man keine EU-Baumusterprüfung mit Bescheinigung durchlaufen muss sondern dass eine Prüfung über die Technische Dokumentation ausreichend ist (für Klasse IIa Produkte).
Besten Dank!
Mit freundlichen Grüßen
Florian Sedlmeir
Sehr geehrter Herr Sedelmeier,
danke für Ihre heutige Frage!
Mit der MDR werden die Karten komplett neu gemischt. D.h. Sie können (und müssen) nun völlig neu entscheiden, welches Konformitätsbewertungsverfahren sie durchlaufen wollen. Auch wenn es Parallelen zu den Verfahren der MDD gibt, können Sie die Entscheidung völlig unabhängig von der MDD treffen.
Abhängig von der Klasse sind die Anhänge XI oder IX möglich. Eine Baumusterprüfung (Type Examination) ist bei Produkten der Klasse IIa nicht erforderlich, wenn der Hersteller die Product Conformity Verification gemäß Anhang XI Teil A durchläuft.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für den interessanten Beitrag.
Nirgendwo in der MDR finde ich einen Hinweis darauf, wie die technische Dokumentation verfasst und archiviert werden muss. Reicht also eine elektronische Archivierung und ein rein elektronisches Handling der DHR’s vollkommen aus?
Vielen Dank für Ihre Rückmeldung
Sehr geehrte Frau Barsch,
die MDR gibt in der Tat keine Technologie vor. Sie bestimmt aber z.B. die Aufbewahrungsfristen. Dazu gibt es einen eigenen Beitrag.
Daneben gibt es noch die Vorgaben zur Dokumentenlenkung aus der ISO 13485, die Sie ebenfalls beachten sollten.
Eine Pflicht zur Archivierung auf Papier gibt es nicht, elektronisch ist okay.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
„Im Sinne der MDR ist ein Medizinprodukt auch als solches zu kennzeichnen. Dazu verwenden Hersteller in der Regel das MD-Symbol. Dieses Symbol ist jedoch erst für die nächste Version der ISO 15223-1 angedacht.
Sofern das MD-Symbol oder andere Symbole verwendet werden, die nicht in der ISO 15223-1:2016 abgebildet sind, dürfen diese zwar benutzt werden, müssen jedoch z.B. in der Gebrauchsanweisung erklärt werden.“
Ist diese Aussage auch zutreffend, wenn der Hersteller noch nicht MDR zertifiziert ist?
Mit freundlichen Grüßen
K. Demir
Sehr geehrte/r K. Demir,
die Anforderungen an die Kennzeichnung gelten für alle Medizinprodukte unabhängig vom Konformitätsbewertungsverfahren.
Die „Zertifizierung“ bezieht sich auf die Anhänge der MDR z.B. eine Zertifizierung für ein Konformitätsbewertungsverfahren nach Anhang IX (QM-System).
Produkte der Klasse I* und höher dürfen Sie nur in den Verkehr bringen, wenn ein Zertifikat vorliegt.
Viele Grüße, Christian Johner
Hallo Herr Professor Johner,
in einem Artikel von 2015 zur Zulassung von Medizinprodukten schreiben Sie, dass es sich bei Medizinprodukten eigentlich nicht um eine Zulassung, sondern um das Durchlaufen eines Konformitätsbwertungsverfahren handelt.
Oben im Text wird allerdings auch der Begriff „Zulassung“ verwendet. Hat sich dies jetzt auch mit der MDR verändert und es handelt sich jetzt um ein richtiges Zulassungsverfahren?
Vielen Dank für Ihre Hilfe!
Sehr geehrte Frau Strohm,
Sie haben absolut Recht! Den Begriff „Zulassung“ nutzt man umgangssprachlich — wie Sie gesehen haben auch ich. Die korrekte Beschreibung wäre die mit den Konformitätsbewertungsverfahren.
Auch wenn Überwachung und Überprüfung der Hersteller bzw. Produkte durch die Benannten Stellen durch die MDR nochmals zugenommen hat, gibt es trotzdem keine Zulassung wie z.B. durch die FDA oder durch die EMA im Fall von Medikamenten.
Viele Grüße, Christian Johner
Sehr geehrte Damen und Herren,
wie verhält es sich mit Geräten, die vor 1995 erstmalig Inverkehr gebracht wurden, d.h. in der Zeit, wo das Inverkehrbringen noch ohne Konformitätsbewertung möglich, d.h. nicht rechtswidrig war. Dürfen diese Geräte jetzt weiter veräußert und bei einem neuen Betreiber betrieben werden (ohne Anwendung der MDR)? Das Inverkehrbringen war bspw. 1993 ohne CE Kennzeichnung erlaubt und nach der damaligen Rechtslage zum Inverkehrbringen wurde das Gerät für sicher befunden. Kann man sich bei heutiger Wiederinbetriebnahme an einem anderen Standort noch darauf berufen?
Mit freundlichen Grüßen
Dorothea Hammer
Sehr geehrte Frau Hammer,
Ihre Frage lässt sich leider nicht so einfach beantworten ohne eine detaillierte Analyse des Sachverhalts. Generell ist das Gerät bereits in Verkehr gebracht und bereits in Betrieb genommen. Somit müsste keine MDR-Konformität erklärt werden, wenn keine neue Inverkehrbringung oder Neuaufbereitung stattfindet vor der Wiederinbetriebnahme. Was unter eine Neuaufbereitung fällt, finden Sie in folgendem Guidance-Dokument beispielhaft erklärt: https://www.team-nb.org//wp-content/uploads/2015/05/nbmeddocuments/Recommendation-NB-MED-2_1-5_rev5_Placing_on_the_market_of_fully_refurbished_medical_devices.pdf
Zu bedenken wäre auch die angegebene Lebenszeit des Geräts bzw. ob diese bereits überschritten ist und vom Hersteller eine weitere Nutzung untersagt ist.
Freundliche Grüße
Luca Salvatore
Sehr geehrter Herr Prof. Dr. Johner,
ich habe eine Frage zum Verkauf eines medizinischen Verbrauchsmaterial in Bezug auf die Verpackungseinheit –
was muss ich als Händler beachten ? Zum Beispiel das Produkt Venenverweilkanüle (steril) in einer VE mit
50 Stück. Diese konnten bisher auch in kleineren Mengen bis hin zu einzeln weiterverkauft werden wenn z.B. ein Kunde diese nur benötigt hatte um seinen Notfallkoffer zu bestücken.
Kann dies weiterhin praktiziert werden solange gesichert ist, dass das Sterilgut in einer 2.ten Umverpackung und der Beilage der Herstellergebrauchsinformation als Kopie abgegeben wird ohne dass man als Händler jetzt zum Hersteller
wird ?
Mit freundlichen Grüßen
Hans-Jürgen Illner
Sehr geehrter Herr Illner,
das ist eine spannende Frage. Sie lässt sich allerdings ohne weitere Informationen nicht ohne weiteres beantworten. Daher ein paar grundlegenden Gedanken:
Eine VE aus einem Produkt herauszuverkaufen, war schon unter der MDD nicht immer erlaubt. Denn der Hersteller bringt das Produkt in den Verkehr. Er hat im Risikomanagement die Risiken für dieses Produkt betrachtet und entsprechende Maßnahmen ergriffen. Beispielsweise könnte eine Maßnahme in Art, Form und Beschriftung der Produktverpackung bestanden haben. Wenn einzelne VEs weiterverkauft werden, dann wäre die Wirksamkeit dieser Maßnahmen nicht mehr sichergestellt.
Genau die gleichen Überlegungen gelten auch für die MDR. Hier kommt allerdings hinzu, dass durch die Änderung der Verpackung – und die Wegnahme einer solchen zählt dazu – der Händler Herstellerpflichten erfüllen müsste. Den Ausnahmetatbestand des Artikels 16 Absatz 2 Unterabsatz b sehe ich nicht als erfüllt.
In der Hoffnung, damit etwas geholfen zu haben, und mit vielen Grüßen, Christian Johner
Die Regel 11 der MDR hemmt ungemein die Neuentwicklung von low risk DiGas und Medical Apps in der EU. Betroffene Unternehmen leiden massiv unter dem Druck Signifikante änderungen an MDD Klasse I Software durchzuführen oder gar eine Neuentwicklung nach MDR durchzuführen. Es ist keine Besserung in Aussicht und führt jetzt bereits dazu, dass Unternehmen den Europäischen Markt ausschließen müssen da eine Weiterentwicklung mit Benannten Stellen unmöglich ist.
Gibt es hier einen Funken Hoffnung auf einen Risikobasierten Ansatz oder sind die Autoren der MDR wirklich gewillt diesen Gesunden Ast der Innovation komplett abzusägen?
Sehr geehrter Herr Docht,
danke für Ihre Frage, die ich auch teilweise als einen Kommentar verstehe.
Dass die MDR die Innovation nicht im Fous hat, sehe ich auch so.
Große Hoffnung, dass wir zu einer Klasse-I für SW zurückkehren habe ich nicht. Denn diese Produkte stärker an die Kandare zu nehmen, ist bewusst erfolgt. Allerdings gibt es mit der MDCG 2019-11 eine gute Argumentationsgrundlage, um nicht in unnötig hohe Klassen zu geraten. Das Klasse-I-Problem löst diese Leitlinie aber nicht.
Beste Grüße, Christian Johner
Ein kleiner technischer Hinweis: Im Text sind einige Hyperlinks auf definierte Begriffe falsch gesetzt.
Beispiel aus Artikel 10:
„The quality management system shall address at least the following aspects:“
Dort liegt unter „system“ ein Link auf die Definition von „System“ (im Sinne von Systeme und Behandlungseinheiten).
Herzliche Grüße
Frank Müller
Vielen Dank, lieber Herr Müller!
Sie haben absolut Recht! Da hat unser Automatismus falsch zugeschlagen. Das ist nicht der richtige Link.
Ich werde mein Team bitten, dass jemand die MDR nochmals durchgeht und die Links prüft.
Nochmals vielen Dank, lieber Herr Müller! Damit helfen Sie, dieses Dokument zu verbessern!
Viele Grüße, Christian Johner
Lieber Herr Johner,
gerne geschehen. Danke Ihnen für die kostenfreie Bereitstellung dieser großartigen, einfach durchsuchbaren Version der MDR.
Herzliche Grüße
Frank Müller
Guten Morgen Herr Prof. Johner,
kann der Fall einer „Neuaufbereitung n. MDR“ nach einer Reparatur (Instandsetzung) eines chirurgisches Instruments (z.B. Schere, Pinzette, Nadelhalter) auftreten?
Danke für Ihre Antwort,
Beste Grüße, Daniela Weber
Sehr geehrte Frau Weber,
die Neuaufbereitung ist „die vollständige Rekonstruktion eines bereits in Verkehr gebrachten oder in Betrieb genommenen Produkts“. Die MDR unterscheidet (aber definiert nicht) den Begriff der Instandhaltung. Daher sind das erst einmal zwei verschiedene Konzepte. Beachten Sie auch den Artikel zur Instandhaltung.
Die Grenze sind aber fließend. Wenn ein chirurgisches Instrument in einem so schlechten Zustand ist, dass es einer vollständigen Rekonstruktion bedarf, würde das unter „Neuaufbereitung“ fallen. Das dürfte aber schon aus Kostengründen eher die Ausnahme sein.
Viele Grüße
Christian Johner
Sehr geehrte Damen und Herren,
gibt es zum Thema MDR-Zertifizierung mittlerweile Wissen oder Erfahrung, welche Auswirkungen es hat, wenn die Benannte Stelle während eines laufenden MDR-Zertifizierungsverfahrens gewechselt wird, der laufende Vertrag also gekündigt wird? Gibt dies (nachteilige) Eintragungen in der EUDAMED, zählt es als ein Fehlversuch etc.? Ich kann hierzu nirgends etwas finden und würde mich sehr freuen, wenn Sie dazu etwas sagen können.
Mit freundlichen Grüßen
Manuela Hartmann
Guten Morgen Frau Hartmann,
prinzipiell ist es auch während der Übergangsfristen möglich, die Benannte Stelle im laufenden Zertifizierungsverfahren zu wechseln und dennoch von den Übergangsfristen zu profitieren. Dafür müssen Sie allerdings die bekannten Voraussetzungen (u.a. QMS nach MDR bis 26.05.2024) erfüllen. Falls der Vertrag beendet wurde aufgrund einer Nichterfüllung von MDR-Anforderungen, gelten die Übergangsfristen nicht mehr. Näheres hierzu finden Sie im Q&A der Kommission in Frage 9.1. „What happens if the application is withdrawn or the written agreement terminated?“ (https://health.ec.europa.eu/system/files/2023-07/mdr_proposal_extension-q-n-a.pdf).
Freundliche Grüße
Luca Salvatore
Liebe Foren-Gemeinschaft,
angestellt bei einem Medizinprodukthersteller, der nunmehr legacy devices der Klasse III als MDR-Produkt transferieren möchte, haben wir folgende Situation. Bei den Klasse III-Produkten handelt es sich um klassische einfache Medizinprodukte auf Verbandstoffbasis. Hier wird sich weder etwas bei Zweckbestimmung noch beim eigentlichen Produktdesign, den Rohstoffen und Lieferanten ändern. Es bleibt quasi alles so wie es seit 30 Jahren ist. Es stellt sich uns nun die Frage, in welchem Umfang überhaupt jetzt eine klassische „Entwicklung“ unter MDR dargelegt werden muss und wie dieses dokumentiert werden muss. Ob es nun ausreicht, darzulegen, dass der wir als Hersteller jegliche Schritte neu bewertet haben und zum Entschluß kommen, dass die bisherige TD unter 93/42 weiterhin als Grundlage genutzt werden, um diese auf MDR zu transferieren. Natürlich unter Beachtung von ggf. erweiterten Anforderungen. Gibt es möglicherweise hierzu schon Erfahrung anderer langjähriger Hersteller.
Mit besten Grüßen
Liebes Johner-Team,
da bei einem unserer MDD-Medizinprodukte das ABS-Material nicht mehr lieferbar ist wollen wir zu einem anderen Material (Cycoloy CX2244ME) wechseln. Allerdings steht im MDS vom Hersteller folgendes „Einige unserer oben genannten Produkte enthalten Zusatzstoffe oder deren Rohstoffe, die auf Materialien tierischen Ursprungs (Schaf- oder Rindergewebe) basieren. Diese Zusatzstoffe oder ihre Rohstoffe, die aus Schafen oder Rindern gewonnen werden (Fettsäuren, Fettalkohole Alkohole, Metallseifen, Fettamine, Fettamide, Fettsäureester, Glycerin usw.) werden normalerweise als Schmiermittel, Gleitmittel, Antistatikum, Emulgator, Antioxidationsmittel oder Korrosionsschutzmittel in Kunststoffe eingearbeitet.“
Dies ist doch für die MDR relevant, oder? Wenn wir auf dieses Material wechseln würden wäre das doch bestimmt eine signifikante Änderung und unser Medizinprdukt könnte nicht mehr unter der MDD laufen, oder? Und müssten wir das Medizinprodukt dann auch kennzeichnen, dass es tierische Materialien enthält?
Vielen Dank im Voraus für Ihre Rückmeldung 🙂
Guten Tag,
Ihre Frage lässt sich schwer pauschal beantworten und weitere Informationen (u.a. Produkt, Patientenkontakt ja/nein/Art des Kontakts). Auch scheint die Aussage im MDS recht pauschal ohne konkrete Referenz auf das konkrete Produkt. Generell sagt das MDCG im Dokument 2020-3, dass es sich im Falle eines „Change to or addition of a new material of human/animal origin“ um eine signifikante Änderung handelt. Ich würde allerdings empfehlen, dies im Einzelfall zu bewerten, basierend auf den genannten Punkten und dem Risikoprofil. Falls Sie bei dieser Bewertung Unterstützung benötigen, dann melden Sie sich gerne bei uns.
Herzliche Grüße
Luca Salvatore