
Wann zählt ein PC zur Medical IT, wann als Medizinprodukt? Die Antwort auf diese Fragen hat weitgehende regulatorische Konsequenzen.
Beispiele für Medical IT
In der Medizintechnik kommen IT-Produkte wie PCs, Monitore oder Switche zum Übertragen, Anzeigen und Speichern (medizinischer) Daten zum Einsatz. Manchmal verwenden die Betreiber diese IT-Produkte alleine, manchmal in Kombination mit Medizinprodukten.
Viele Hersteller versehen diese IT-Produkte mit dem Begriff „Medical“:
- „Medical PC“ = PC zum Einsatz in der Medizin(-technik)
- „Medical LCD“ = LC-Monitor zum Einsatz in der Medizin und Medizintechnik
- „Medical Switch“ = Switch zum Übertragen medizinischer Daten
Weitere Beispiele für Medical-IT-Produkte sind speziell für den medizinischen Bereich vorgesehene Trenntransformatoren, Signal-Trennverstärker wie Video-Trennverstärker und USVs (Unabhängige Spannungsversorgungen).
Der Zusatz „Medical“ bei diesen Medical IT Produkten dient häufig auch Marketing-Zwecken.

Medical IT: Auch ein Medizinprodukt?
Was unter Medical-IT zu verstehen ist
Der Begriff „Medical IT“ ist in keiner Europäischen Harmonisierungs-Richtlinie (etwa der Medizinprodukterichtlinie) definiert. Man findet ihn auch nicht in deutschen Gesetzen wie dem Medizinproduktegesetz (MPG) oder Produktsicherheitsgesetz (ProdSG). Diese Gesetze setzen die entsprechenden EU-Richtlinien in deutsche Gesetze um.
Das Attribut „Medical“ soll dem potenziellen Käufer signalisieren, dass das IT-Produkt für einen Einsatz in der Medizin bzw. Medizintechnik vorgesehen und dafür in besonderer Weise geeignet ist.
Aufgrund einer fehlenden regulatorischen Definition des Begriffes „Medical IT-Produkt“ werden derartige Produkte gelegentlich als „Medizinprodukt“ missverstanden und fälschlicherweise auch so beworben.
Das liegt auch daran, dass die Hersteller sogar in den Unterlagen und Marketinginformationen nicht immer klar beschreiben, nach welchen EU-Richtlinien bzw. gemäß welchen gesetzlichen Anforderungen sie die CE-Kennzeichnung an ihre Produkte anbringen und sie somit in Verkehr bringen.
Daher wird nachfolgende Definition für ein mit dem Zusatz „Medical“ versehenes IT-Produkt vorgeschlagen.
Ein IT-Produkt, das der Hersteller für den Einsatz im medizinischen Bereich vorsieht, das aber selbst kein Medizinprodukt ist, jedoch nachweislich die Anforderungen des deutschen Produktsicherheitsgesetzes (ProdSG), des deutschen EMV-Gesetzes (zur elektromagnetischen Verträglichkeit) sowie medizintechnischer Normen erfüllt.
Der Marketing-Begriff „Medical“ ist also nur dann gerechtfertigt, wenn die vorstehenden Anforderungen nachweislich erfüllt sind. Ein Medical-IT-Produkt stellt somit kein Medizinprodukt gemäß Medizinprodukte-Richtlinie bzw. Medizinproduktegesetz dar, auch wenn der Begriff „Medical“ häufig unzulässigerweise so verwendet und auch verstanden wird.
Regulatorische Anforderungen an Medical-IT-Produkte
ProdSG und EMVG
Während das Produktsicherheitsgesetz u. a. die europäische Niederspannungs-Richtlinie in deutsches Recht für technische Produkte und Verbraucherprodukte umsetzt, beinhaltet das Gesetz über die elektromagnetische Verträglichkeit von Betriebsmitteln (EMVG) die Anforderungen der europäischen EMV-Richtlinie.
Um ein IT-Produkt im Bereich der Medizin und Medizintechnik, insbesondere in der sogenannten Patientenumgebung, zulässigerweise einzusetzen, kann und ggf. muss es ein Hersteller entsprechend den Anforderungen der folgenden Normen der Medizintechnik bauen lassen:
- Elektrische Sicherheit (Safety): DIN EN 60601-1: Medizinische elektrische Geräte Teil 1-1: Allgemeine Festlegungen für die Sicherheit – Ergänzungsnorm: Festlegungen für die Sicherheit von medizinischen elektrischen Systemen und
- Elektromagnetische Verträglichkeit (EMV): DIN EN 60601-1-2: Medizinische elektrische Geräte Teil 1-1: Allgemeine Festlegungen für die Sicherheit einschließlich der wesentlichen Leistungsmerkmale – Ergänzungsnorm: Elektromagnetische Störgrößen – Anforderungen und Prüfungen
Diese Normen enthalten u.a. engere und schärfere Grenzwerte und Bestimmungen für Ableitströme und EMV, als sie für gewöhnliche IT-Produkte gelten.
Ein Hersteller eines Medical-IT-Produktes weist durch ein entsprechendes Zertifikat einer anerkannten Prüfstelle nach, dass die Anforderungen dieser beiden medizintechnischen Normen für das Produkt erfüllt sind.
Ein Medical-IT-Produkt erhält nach Durchführung des Konformitätsbewertungsverfahrens zwar eine CE-Kennzeichnung gemäß diesen beiden Richtlinien, stellt aber kein Medizinprodukt nach Medizinprodukte-Richtlinie dar.
IEC 60601-1 oder IEC 60950?
Wenn ein Medical IT-Produkt in der Patientenumgebung eingesetzt werden soll (die Zweckbestimmung des Herstellers muss das vorsehen), dann muss das Produkt den Anforderungen der 60601-1 entsprechen. Wenn es hingegen laut Zweckbestimmung außerhalb der Patientenumgebung eingesetzt werden soll, genügt die Konformität mit der IEC 60950.
Das Dilemma mit diesen Produkten besteht oft darin, dass die Betreiber sie (entgegen der Zweckbestimmung) doch in der Patientenumgebung einsetzen. Dann ist der Betreiber in der Pflicht, das Kapitel 16 der 60601-1 zu beachten (siehe § 2 Ab. 1 der MPBetreibV). Aber diese Pflicht wird häufig nicht erfüllt.
Daher empfehlen wir, solche Produkte von vorn herein konform den Anforderungen der IEC 60601-1 zu bauen und dies nachzuweisen.
Eine Herausforderung dabei besteht darin, dass die IEC 60601-1 ein Risikomanagement bedingt. Damit sind Firmen, die sonst keine Medizinprodukte herstellen sehr gefordert. Daher beschränken sich die Prüfungen meist auf die Basissicherheit und weniger auf die wesentlichen Leistungsmerkmale.
Weitere Anforderungen
Ein Medical IT-Produkt weist für den Einsatz in der Medizin/Medizintechnik auch eine IPX-Klassifikation (IP = Ingress Protection) als Schutz gegen das Eindringen von Feuchtigkeit und Flüssigkeit auf.
Zusätzlich muss ein solches Medical IT-Produkt auch hygienische Anforderungen erfüllen, z. B. die RKI-Richtlinie, um für den Einsatz unter besonderen Anforderungen wie in der Medizin bzw. Medizintechnik geeignet zu sein. Dazu gehören u. a. möglichst glatte und leicht zu reinigende Flächen und Nachweise zur Desinfizierbarkeit (Beständigkeit gegen Standard-Mittel). Achtung: Ein Hinweis auf die Verwendung von „Standard-Desinfektionsmitteln“ reicht NICHT aus.
Aufgaben der Hersteller
Schritt 1: Produkt klassifizieren
Wenn ein Hersteller ein technisches Produkt zur Verwendung in der Medizin bzw. Medizintechnik in Verkehr bringen will, muss er sich gemäß Abbildung 1 entscheiden, ob es sich gemäß der von ihm festzulegenden Zweckbestimmung um ein Medizinprodukt nach Medizinprodukte-Richtlinie oder aber um ein technisches Produkt nach Niederspannungs- und EMV-Richtlinie handelt, also um ein Medical-IT-Produkt.
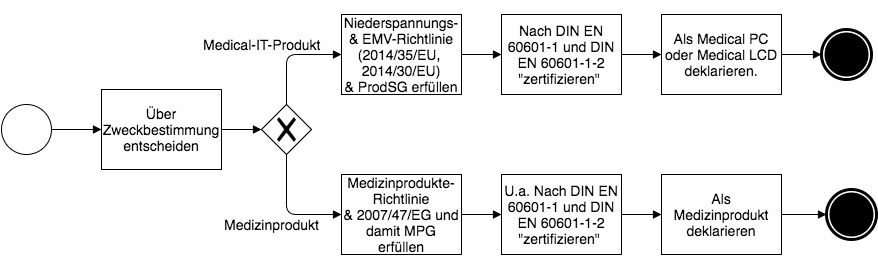
Abb. 1: Entscheidung über die regulatorische Zuordnung (zum Vergrößern klicken)
Schritt 2: Regulatorische Anforderungen erfüllen
Wenn der Hersteller sein IT-Produkt – z.B. PC, LC-Monitor oder andere IT-Komponenten – für den Einsatz in der Medizin/Medizintechnik z. B. in Kombination mit Medizinprodukten vorsieht, kann und muss er die o.g. regulatorischen Anforderungen umsetzen. Mit dieser konstruktiven Umsetzung ermöglicht er den Einsatz der betreffenden IT-Produkte in der Medizin und Medizintechnik.
Schritt 3: Konformität erklären
Es steht jedem Hersteller frei, für sein IT-Produkt (nach Niederspannungs- und EMV-Richtlinie) zusätzlich die Anforderungen der beiden medizintechnischen Normen umzusetzen und mit einem zusätzlichen Prüfbericht oder -zertifikat nachzuweisen. Er muss das nur tun, wenn er diese Produkte für den Einsatz in der Medizin bzw. Medizintechnik vorsieht. Der Nachweis der Erfüllung wird mit einer Technischen Dokumentation, auch als Produkthauptakte bezeichnet, belegt.
Erst, wenn er die beiden Normen erfüllt, ist der Begriff „Medical IT-Komponente“ wie in „Medical“ LC-Monitor oder „Medical“ PC gerechtfertigt. Ein solcher Monitor unterscheidet sich dann vom Büromonitor durch medizingerätespezifische Eigenschaften, z. B. durch geringere Ableitströme nach DIN EN 60601-1 wie den Gehäuseableitstrom oder physiologisch orientierte EMV Prüfungen nach DIN EN 60601-1-2 und in der Hygiene.
Erst dann darf der Hersteller oder sein Bevollmächtigter mit Sitz in der EU die Übereinstimmung des Produktes mit diesen Anforderungen mittels der EU-Konformitätserklärung bestätigen und die CE-Kennzeichnung anbringen.
Nochmals: Abgrenzung Medical IT und Medizinprodukt
Aber: Die alleinige Erfüllung der beiden genannten medizintechnischen Normen reicht nicht aus, um die Konformität mit der Medizinprodukterichtlinie nachzuweisen bzw. zu erklären! Ein PC, der nach den Anforderungen der IT-Norm DIN EN IEC 60950 gebaut wird und zusätzlich die Anforderungen nach DIN EN IEC 60601-1 und DIN EN IEC 60601-1-2 erfüllt, bleibt dennoch ein Nicht-Medizinprodukt, ein Medical-IT-Produkt! Ein solcher PC darf aber in der Patientenumgebung eingesetzt werden, weil er die Anforderungen der vorgenannten Normen erfüllt.
Aufgaben der Betreiber
Da in der Praxis die Bezeichnung Medical-IT-Produkt häufig als Synonym für Medizinprodukt verstanden oder aber gleich gesetzt wird, werden immer wieder solche IT-Produkte beschafft, die für den vorgesehenen Einsatz in der Medizin bzw. Medizintechnik entweder nicht geeignet sind (da sie die normativen Anforderungen der Medizintechnik-Normen nicht erfüllen) oder nicht korrekt nach den gesetzlichen Anforderungen des MPG bzw. des ProdSG in Verkehr gebracht wurden.
Schritt 1: Anwendungszweck festlegen
Daher muss der Betreiber, i.d.R. vertreten durch die Fachabteilungen IT und Medizintechnik, zunächst vor der Beschaffung beschreiben und festlegen, für welchen Anwendungszweck und unter welchen Voraussetzungen IT-Komponenten in der Medizin unter Beachtung der Medizinprodukte-Betreiberverordnung und der technischen Normen eingesetzt werden sollen.
Folgende Beispiele verdeutlichen die Anforderungen am Beispiel eines LC-Monitors:
- LC-Monitor für den Aufruf von KIS und anderen IT-Applikationen in Funktionsbereichen und OP außerhalb der Patientenumgebung unter Beachtung der VDE 0100 Teil 710 = herkömmliches IT-Produkt
- LC-Monitor für den Aufruf von KIS und anderen IT-Applikationen in Funktionsbereichen und OP innerhalb der Patientenumgebung = „Medical“ LCD
- LC-Monitor für die Visualisierung medizinischer Bilder (Farbinformation z. B. für die Endoskopie, Ableitung von Diagnose wie z. B. in der Befundung von Röntgenbildern) innerhalb und außerhalb der Patientenumgebung = Medizinprodukt LC-Monitor.
Schritt 2: Produkt beschaffen und Konformitätsvermutung prüfen
Wenn also IT-Produkte für die Medizintechnik beschafft werden sollen, sollte sich der Betreiber die erforderlichen Unterlagen und Dokumente, insbesondere die Zweckbestimmung, die Konformitätserklärung und die Prüfberichte für „Medical“ IT-Produkte vorlegen lassen.
Nicht immer sind die Herstellerunterlagen für Medical IT-Komponenten eindeutig und vollständig. Der Betreiber, vertreten durch die Fachabteilungen IT und/oder Medizintechnik, hat eine Prüfpflicht bei der Beschaffung, auch bei IT-Produkten sowie bei der Beschaffung von Medizinprodukten, die sich aus der Medizinprodukte-Betreiberverordnung heraus ergibt.
Schritt 3: Bei Abweichungen oder im Zweifelsfall reagieren
Stellt der Betreiber bei der Überprüfung fest oder hat er Zweifel, ob die vorgelegten Unterlagen vollständig und/oder korrekt das Inverkehrbringen nach EU-Richtlinnen bestätigen, bedeutet dies im regulatorischen Sinne, dass die sogenannte Vermutungswirkung aufgehoben wird. In einem solchen Fall sollte sich der Betreiber an die zuständigen Marktaufsichtsbehörden wenden, um klären zu lassen, ob ein Produkt ordnungsgemäß in Verkehr gebracht wurde und/oder die Anforderungen der zutreffenden Richtlinie(n) erfüllt sind oder nicht.
Im Zweifelsfall übernimmt sonst der Betreiber, also z.B. ein Krankenhaus, die regulatorische Verantwortung für ein vom Hersteller nicht ordnungsgemäß in Verkehr gebrachtes Produkt. Das Krankenhaus ist dann Inverkehrbringer eines solchen Produktes und übernimmt damit im Schadensfall auch die Haftung für ein solches Produkt.
Es finden sich immer wieder Beispiele wie die folgenden, die zeigen, dass die genannten regulatorischen Anforderungen nicht erfüllt sind:
Beispiele für Nicht-Konformitäten
Beispiel 1: Fehlende oder unvollständige Konformitätserklärung
Ein Typenschild eines „Medical“ PC ist mit dem Symbol „CE“ versehen. In den Unterlagen des Produktes fehlt die Konformitätserklärung, aus der hervorgehen muss, nach welcher Richtlinie bzw. welchen Richtlinien das Produkt in Verkehr gebracht wurde. Der Hersteller bezeichnet das Produkt als Medizinprodukt der Klasse I.
Auf Grund von Nachfragen stellt sich heraus, dass es sich nicht um ein Medizinprodukt handelt, sondern die Angabe der Klasse I sich auf die Schutzklasse des Produktes [Anm.: gemäß IEC 60601-1 – elektrische Sicherheit] bezieht.
Das Produkt ist nicht ordnungsgemäß in Verkehr gebracht, weil die Konformitätserklärung fehlt, die nach JEDER Richtlinie erforderlich ist. Zusätzlich kann kein Nachweis über eine Technische Dokumentation für eine Konformitätsbewertung vorgelegt werden.
Hinweis: Datenblätter zu einem solchen Produkte oder zu den verwendeten Teilkomponenten, aus denen eine CE-Kennzeichnung und/oder eine Prüfung nach einer spezifischen Norm hervorgehen, reichen zu diesem Zweck NICHT aus.
Beispiel 2: Unvollständige Abdeckung der Normen
In den Unterlagen eines „Medical“ Produktes finden sich Angaben, dass das Produkt nur nach der IEC 60601-1 geprüft wurde. Eine Prüfung der medizinischen EMV-Norm IEC 60601-1-2 ist nicht nachgewiesen.
Beispiel 3: Falsche Klassifizierung des Produkts
Ein Hersteller weist in der Konformitätserklärung aus, dass sein Produkt den Anforderungen der Medizinprodukte-Richtlinie, Niederspannungs-Richtlinie und EMV-Richtlinie entspricht.
Wenn ein Hersteller die Konformität nach den drei verschiedenen Richtlinien (Niederspannungs-, EMV- und Medizinprodukterichtlinie) erklärt, dann ist dies nicht korrekt. Entweder gelten für ein solches Produkt Niederspannungs- und EMV-Richtlinie oder die Medizinprodukterichtlinie, die wesentlich enger gefasste Grenzwerte für die elektrische Sicherheit und für die EMV (Emission und Immission) für Medizinprodukte aufweist und die Anwendbarkeit der anderen beiden Richtlinien ausschließt.
Die Medizinprodukte-Richtlinie enthält im Einleitungsteil der Erwägungsgründe folgenden klaren Hinweis:
Die Gesichtspunkte der elektromagnetischen Verträglichkeit sind wesentlicher Bestandteil der Unbedenklichkeit der Medizinprodukte. Die vorliegende Richtlinie enthält gegenüber der Richtlinie 89/336/EWG des Rates vom 3. Mai 1989 zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über die elektromagnetische Verträglichkeit entsprechende spezifische Bestimmungen.
Mit anderen Worten: für ein Medizinprodukt findet die EMV-Richtlinie (aktuell 2014/30/EU) grundsätzlich keine Anwendung, da die Medizinprodukte-Richtlinie spezifische Anforderungen an EMV-Schutz von Medizinprodukten enthält. In Analogie gilt für Medizinprodukte auch nicht die Niederspannungs-Richtlinie.
Beispiel 4: Verwendung nicht definierter Begriffe
Der Hersteller verwendet in seinen Materialien Begriffe wie
- CE-Zulassung
- CE-Prüfung
- CE-Zertifizierung
- MPG-Prüfung
- MPG-Zulassung
- Medizinzulassung
- Medizinische Zulassung
So etwas wäre nicht korrekt, da die Begriffe in keiner einzigen EU-Rechtsvorschrift definiert sind. Derartige Begriffe lassen vermuten, dass die rechtlich geforderten Konformitätsanforderungen der Harmonisierungsrechtsvorschriften für ein solches Produkt nicht korrekt umgesetzt wurden. Die Erwähnung solcher Begriffe sollte den potenziellen Käufer zu Vorsicht bzw. zu intensivem Nachfragen veranlassen!
Empfehlungen
Damit Krankenhäuser und ihre IT-Abteilungen ihre Medizintechnik und IT-Produkte zum Einsatz in der Medizin/Medizintechnik ordnungsgemäß beschaffen und somit haftungsrechtliche Probleme mit solchen Produkten vermeiden können, sollten sie in Anfragen und Ausschreibungstexten den vorgesehenen Verwendungszweck und die regulatorischen Anforderungen (Medizinprodukt, IT-Produkt) eindeutig beschreiben und abfragen.
Anfragen und Ausschreibungstexte sollten klar beinhalten, dass folgende Informationen und Unterlagen vorgelegt werden, damit ein Krankenhaus eine ordnungsgemäße Prüfung vornehmen kann:
- Zweckbestimmung
- Aktuelle Konformitätserklärung
- Prüfbescheinigungen über die Prüfung nach DIN EN 60601-1 und DIN EN 60601-1-2
Sind die Anforderungen nicht klar definiert, wird es einem Krankenhaus schwer fallen, von einer Beschaffung von „Medical“ IT-Produkten zurückzutreten, wenn sich herausstellt, dass die beschafften Produkte nicht ordnungsgemäß in Verkehr gebracht wurden und somit eine CE-Kennzeichnung nicht bzw. nicht korrekt angebracht worden ist.



Sehr geehrter Herr Prof. Johner,
kann denn ein Hersteller eines Medical-PCs eine Konformitätserklärung nach der Medizinprodukterichtlinie ausstellen, wenn er in der Dokumentation folgende Zweckbestimmung angibt:
Intended use
XYZ was applied to nursing mobile cart computer.
In der Konformitätserklärung heißt es:
The following products herewith confirmed to comply with Directives of 93/42/EEC and Regulation of 2017/745.
….-Liste der Produkte- …
The listed standards as bellow were applied:
EN 60601-1-2:2015
IEC 60601-1:2005/A1:2012
Zumindest die Erklärung nach MDD und MDR ist m. E. schon falsch. Aber selbst die Konformität nach nur einer Richtlinie scheint mir anhand des „intended use“ nicht richtig.
Vielen Dank im Voraus für Ihre Infos.
Beste Grüße,
W. Neumüller
Sehr geehrter Herr Prof. Johner,
ich bin auf diesen Artikel gestoßen, als ich auf der Suche nach einer Antwort zu einem verwandten Thema war: Wenn wir einen einfachen Monitor zusammen mit unserer Stand-Alone-Software (Medizinprodukt) an eine Klinik liefern, hat das regulatorischen Einfluss auf unser Medizinprodukt, unsere SOPs usw.? Wir haben herausgefunden, dass ein Kunde keinen extra Monitor zur Anzeige der Vorhersagesoftware im Ärztezimmer (also nicht Patientenumgebung) gekauft hat, sondern einen Monitor verwendet, auf dem auch andere Anwendungen genutzt werden, so dass die Ärzte aktiv die Anwendung wechseln müssen, um unsere Software zu sehen – was bedeutet, dass sie sie nicht so oft nutzen, wie wir es uns wünschen würden. Wir würden ihnen also einen Monitor geben. Aber wird dieser dann damit automatisch Teil unseres Medizinprodukts und müssen wir unsere SOP für Tracking und Lieferung erweitern (bisher gibt es nur einen Software-Download, sonst nichts)? Jenseits davon befürchte ich, dass es ein Problem mit dem Heilmittelwerbegesetz ist, wenn wir es ihnen schenken (da es bei diesem bestehenden Kunden nicht in der Rechnung/dem Vertrag enthalten ist, in Zukunft könnte es das aber sein)? Über Ihre Einschätzung würde ich mich sehr freuen.
Liebe Frau Hartmann,
vielen herzlichen Dank für Ihre Frage.
Ich bin noch nicht sicher, ob ich die Fragen (es sind mehrere) richtig versanden habe. Ich versuche es einmal und wenn ich falsch liege, melden Sie sich gern nochmals.
Monitor als Teil vom Produkt:
Ich verstehe unter einem „Monitor“ einen Bildschirm, der zur Anzeige dient. Falls die Software spezifische Anforderungen an Auflösung, Bilddiagonale oder Farbraum hat und deshalb ein Monitor mitgeliefert wird, wird dieser als Bestandteil des Produkts betrachtet. Alternativ können Sie diese Anforderungen auch im Benutzerhandbuch festlegen, und in diesem Fall wird kein Monitor mitgeliefert. Diese Vorgehensweise wäre vor allem angemessen, wenn gängige Standardmonitore verwendet werden können.
Falls spezielle Anforderungen für den Monitor gelten und ein Benutzer die Softwareausgabe auf einem beliebigen Monitor anzeigt und dies zu einer fehlerhaften Darstellung führt, sollten im Benutzerhandbuch die damit verbundenen Risiken deutlich hervorgehoben werden. Wie Sie sehen, müssen Sie diese Aspekte in der Risikoanalyse behandeln und entsprechende Maßnahmen definieren.
Abgabe des Monitors:
Ich kann Ihnen leider nicht sagen, ob das Verschenken des Monitors erlaubt ist, da ich nicht mit den relevanten Werbegesetzen vertraut bin. Allerdings gibt es auch dann keine Gewährleistung dafür, dass der Monitor tatsächlich genutzt wird.
Herzlichst, Mario Klessascheck
Lieber Herr Klessascheck,
vielen Dank für Ihre Antworten! Wir haben keinerlei Vorgaben an den Computerbildschirm/ Monitor, auf dem die Software angezeigt wird, nur eine empfohlene Bildschirmauflösung. Insofern kann ich also davon ausgehen, dass er auch nicht Teil des Produkts wird, wenn wir zukünftig im Vertrag festlegen, dass wir einen mitliefern, richtig? (Ich bin da so vorsichtig, weil ich früher mal ein stoffliches Medizinprodukt hatte und einen Löffel mitliefern wollte zur Entnahme, der leider (von uns nicht gewollt) Striche für 1ml etc. hatte – und damit ein Medizinprodukt war, wodurch das Ganze zum Medizinproduktesystem geworden wäre…).
Und dementsprechend brauche ich auch keine SOP für die Verpackung / Lieferung des Monitors, weil das nix mit dem QM-System zu tun hat?
Bzgl. HWG und anderen Gesetzen bzgl. der Abgabe informiere ich mich dann an anderer Stelle. Vielen Dank!
Liebe Frau Hartmann,
Ich bin mir noch nicht sicher, ob ich verstanden habe, warum Sie einen Monitor vertraglich festlegen möchten, wenn Sie keine besonderen Anforderungen an die Hardware haben. Aus Sicht der Software müssen Sie die Anforderungen an den Computer, an das Betriebssystem und den Bildschirm definieren. Wenn allgemein verfügbare Computer und Bildschirme diese Anforderungen definieren, wäre es ausreichend dies im Benutzerhandbuch zu beschreiben. Wenn Sie den Bildschirm mitsenden und damit nahelegen, dass dieser Bildschirm verwendet werden soll, dann müssten Sie die Sachgesamtheit auch als Produkt betrachten.
Ich habe hier einmal einen weiteren Artikel verlinkt Medizinprodukte-PC: Software-Hersteller, aufgepasst!. Vielleicht hilft der bei der Entscheidung.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
vielen Dank für Ihre Antwort und den Link, beides haben geholfen zu entscheiden, dass wir nichts dergleichen tun werden.
Wir wollten den Kliniken gern den Monitor inkl. PC zur Verfügung stellen, damit sie die Software auch oft und gut nutzen können und vertraglich wollte ich das festlegen, damit wir keine Probleme mit Compliance haben, da sie sowas sonst nicht annehmen können. Aber es scheint alles zu kompliziert und wir lassen es.
Schöne Grüße,
Manuela Hartmann