
Die Medical Device Coordination Group (MDCG) hat ein Guidance Document entworfen, das beschreibt, wie Hersteller ihre Medizinprodukte der Klasse 1 MDR-konform in den Verkehr bringen sollen.
Das Dokument trägt den Titel Guidance Notes for Manufacturers of Class I Medical Devices. Dieser Artikel fasst dieses Dokument zusammen und gibt Herstellern dieser Produkte Tipps.
1. Was Medizinprodukte der Klasse 1 auszeichnet
a) Einteilung von Medizinprodukten in Klassen
Die Medizinprodukteverordnung MDR unterteilt Medizinprodukte in die Klassen 1, 2a, 2b und 3. Genauso macht es die Medizinprodukterichtlinie MDD. Die Klassen werden häufig auch mit römischen Ziffern geschrieben (Klasse I, IIa, IIb und III).
Die Klassifizierungsregeln weisen Produkten mit höheren Risiken höhere Klassen zu.
Beispiele für Medizinprodukte der Klasse I sind OP-Tücher und viele chirurgische Instrumente wie Pinzetten, Latexhandschuhe, Masken, Rollstühle (ohne Motor), Stethoskope, Verbände oder Brillen.
b) Auswirkung der Klassifizierung
Die Klassen haben keinen (!) Einfluss auf die grundlegenden Sicherheits- und Leistungsanforderungen, deren Einhaltung die Hersteller nachweisen müssen. Vielmehr bestimmen die Klassen die Konformitätsbewertungsverfahren, um die Konformität mit ebendiesen Sicherheits- und Leistungsanforderungen nachzuweisen.
Für Medizinprodukte der Klasse 1 müssen keine Benannten Stellen in das Konformitätsbewertungsverfahren einbezogen werden. Zudem besteht die MDR bei Medizinprodukten der Klasse 1 nicht auf der Zertifizierung des Qualitätsmanagementsystems durch eine Benannte Stelle. Beides spart Zeit und Kosten.
Eine Ausnahme bilden Medizinprodukte der Klassen 1s, 1r und 1m:
- 1s: Produkte, die in sterilem Zustand in Verkehr gebracht werden
- 1r: Wiederverwendbare chirurgische Instrumente (r steht für „reusable“)
- 1m: Produkte mit Messfunktion
Für diese „1*-Produkte“ müssen die Hersteller bei der Konformitätsbewertung Benannte Stellen einbinden.
Lesen Sie mehr über die Klassifizierung von Medizinprodukten und über Konformitätsbewertungsverfahren. Der Kategorieartikel „Regulatory Affairs“ verschafft eine Übersicht über alle regulatorischen Vorgaben.
Bitte beachten Sie auch die Hinweise zur Software der Klasse I.
2. Wie Medizinprodukte der Klasse 1 „zugelassen“ werden
Die MDCG beschreibt acht Schritte, die Hersteller bei der Inverkehrbringung durchlaufen sollten.
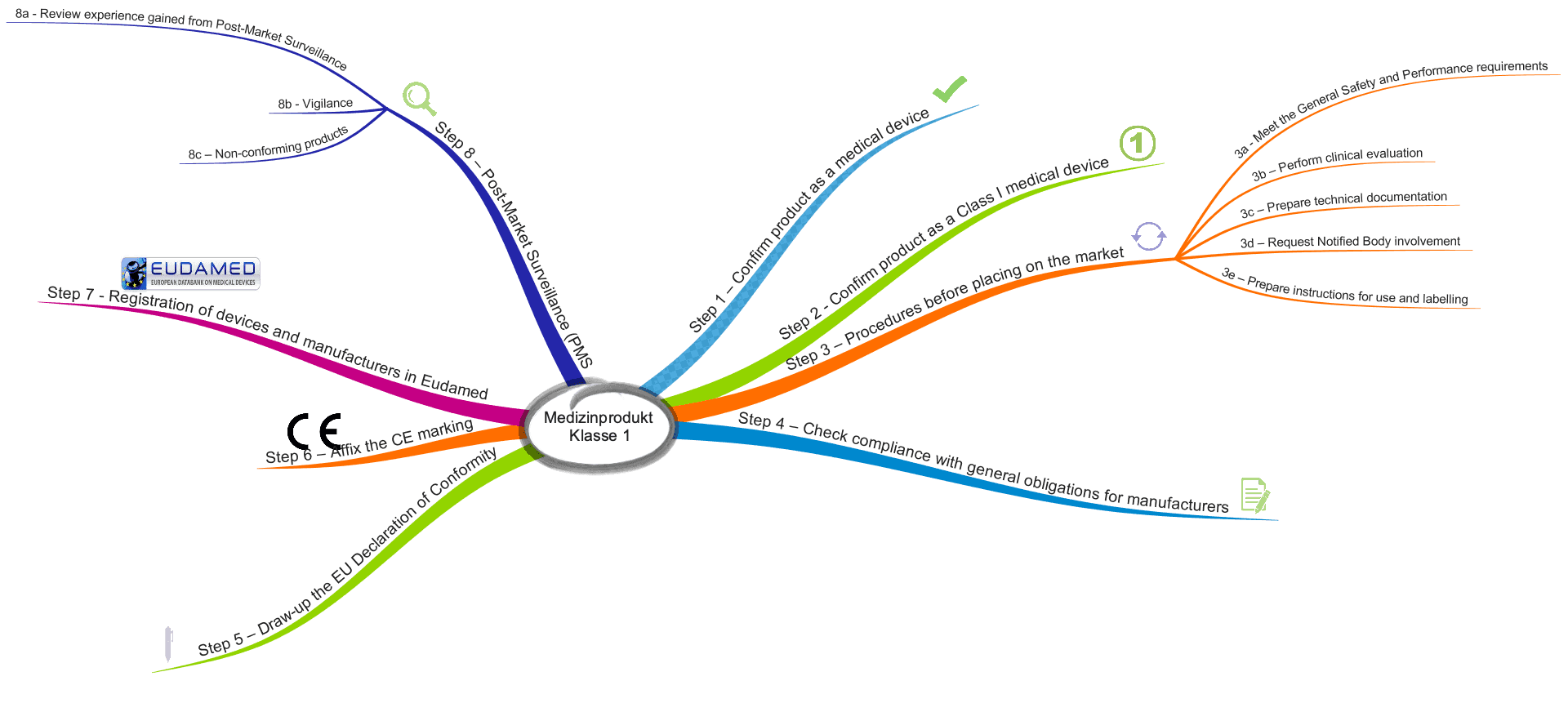
Schritt 1: Überprüfung und Bestätigung, dass das Produkt ein Medizinprodukt ist
Die MDR hat die Definition des Begriffs „Medizinprodukt“ nahezu unverändert aus der MDD übernommen. Daher ist es unwahrscheinlich, dass ein Produkt, das bisher ein Medizinprodukt war, nun nicht mehr unter den Anwendungsbereich der MDR fällt.
Schritt 2: Überprüfung, dass es ein Medizinprodukt der Klasse 1 ist
Dieser Schritt ist deshalb wesentlich, weil die MDR die Klassifizierungsregeln geändert hat. Beispielsweise fällt jetzt nahezu jede Standalone-Software nicht mehr in diese niedrigste Klasse!
Das Guidance-Dokument der MDCG, das dieser Artikel diskutiert, ist nur für Medizinprodukte der Klasse 1 anwendbar, auch wenn die meisten Anforderungen für alle Medizinprodukte gelten.
Schritt 3.a: Sicherstellen, dass die grundlegenden Sicherheits- und Leistungsanforderungen erfüllt sind
Dass die grundlegenden Sicherheits- und Leistungsanforderungen des Anhangs I zu erfüllen sind, ist offensichtlich. Die MDCG weist aber darauf hin, dass ggf. auch andere Vorschriften wie die Maschinenrichtlinie zu beachten sind.
Sie betont die Bedeutung des Risikomanagements und erinnert daran, dass der Nachweis der Anforderungen durch Anwendung harmonisierter Normen und Common Specifications möglich ist.
Schritt 3.b: Klinische Bewertung erstellen
Die MDCG weist darauf hin, wie wichtig der MDR die klinische Bewertung ist und dass die MDR darauf besteht, dass Hersteller
- alternative Behandlungen analysieren und vergleichen,
- Post-Market-Daten berücksichtigen,
- die Akzeptanz des Nutzen-Risiko-Verhältnisses auf Basis klinischer Daten nachweisen und
- klinische Prüfungen durchführen, wenn die vorliegende Datenlage (klinische Daten oder auch Leistungsdaten) nicht ausreichend ist.
Schritt 3.c: Technische Dokumentation erstellen
Hersteller von Medizinprodukten der Klasse 1 müssen die technische Dokumentation konform den Anhängen II und III erstellen. Das gilt natürlich für alle Medizinprodukte.
Das MDCG-Dokument geht auf einige Änderungen ein, die die MDR im Vergleich zur MDD eingeführt hat:
- Begründung der Klassifizierung
- Basis UDI-DI
- Verweis auf Vorgängerprodukte und ähnliche Produkte
- Verweis auf angewendete und dann gültige harmonisierte Normen und „Common Specifications“
- Beschreibung des Post-Market-Surveillance-Systems
Schritt 3.d: Anfrage bei Benannter Stelle
Wie bereits oben erwähnt, müssen Hersteller von Medizinprodukten der Klasse 1* eine Benannte Stelle einbeziehen, die für die entsprechenden Produktklassen akkreditiert ist:
- Devices in sterile condition: Code MDS 1005
- Reusable surgical instruments: Code MDS 1006
- Devices with a measuring function: Code MDS 1010
Hersteller können in der NANDO-Datenbank einsehen, welche Benannten Stellen für den jeweiligen Code akkreditiert sind.
Schritt 3.e: Gebrauchsanweisung und Labeling vorbereiten
Etwas überraschend ist, dass erst im Schritt 3.e die MDCG die Gebrauchsanweisung und das Labeling adressiert. Schließlich bestimmt der Anhang I deren Inhalt. Auch die klinische Bewertung setzt eine Gebrauchsanweisung voraus.
Die MDCG erwähnt, dass für Medizinprodukte der Klasse I keine Gebrauchsanweisung vorgeschrieben ist, wenn die sichere Nutzung gewährleistet ist. Weiter geht das Dokument auf die Sprachen ein (ohne eine Liste mit den geforderten Sprachen zu referenzieren) und auf die Pflicht der Händler, die Begleitmaterialien in diesen Sprachen bereitzustellen.
Symbole werden über harmonisierte Normen und Common Specifications vorgegeben; das Label muss klarstellen, dass das Produkt ein Medizinprodukt ist.
Schritt 4: Prüfen der Konformität mit den Anforderungen an die Hersteller
In diesem vierten Schritt geht es weniger um das Produkt als um den Hersteller, v. a. um dessen Pflicht, ein QM-System zu etablieren und eine Versicherung abzuschließen.
Auf die neue Pflicht nach einer verantwortlichen Person gemäß Artikel 15 geht die MDCG nicht an dieser Stelle ein, sondern bereits in der Einleitung.
Schritt 5: Konformitätserklärung erstellen
Wenig überraschend ist die Forderung, dass Hersteller die Konformität mit der MDR und ggf. weiteren EU-Vorschriften erklären und diese Konformitätserklärung in die Landessprachen übersetzen müssen, in denen das Produkt angeboten wird.
Schritt 6: CE-Kennzeichnung anbringen
Dieser Schritt ist ebenfalls offensichtlich: Auch die Hersteller von Medizinprodukten der Klasse I müssen die CE-Kennzeichnung anbringen, wobei bei Klasse-1*-Produkten die Nummer der Benannten Stelle Teil dieser Kennzeichnung sein muss.
Schritt 7: Registrierung des Produkts und des Herstellers in der EUDAMED
Der siebte Schritt ist in dieser Form neu: Hersteller sind verpflichtet, sich in der EUDAMED zu registrieren, woraufhin ihnen eine „SRN“ zugeteilt wird. Das Gleiche gilt übrigens für EU-Repräsentanten und Importeure.
Anschließend ist es die Aufgabe der Hersteller, die Medizinprodukte zu registrieren. Dabei werden UDI-DI und Basis UDI-DI zugeteilt.
Solange die EUDAMED nicht voll funktionsfähig sei, müssen die Hersteller bzw. EU-Repräsentanten die zuständige Behörde (in Deutschland das BfArM) informieren bzw. das Produkt registrieren.
Schritt 8.a: Post-Market-Daten sammeln und bewerten
Die MDR verpflichtet die Hersteller zur Etablierung eines PMS-Systems, das Teil des QM-Systems ist. Für Medizinprodukte der Klasse 1 müssen die Hersteller einen Bericht über die Überwachung nach dem Inverkehrbringen erstellen, den Post-Market Surveillance Report (PMS-Report=. Die Anforderungen an diesen Bericht sind geringer als an die Periodic Safety Update Reports (PSUR).
Schritt 8.b: Vigilanzsystem
Die MDCG erinnert, dass die MDR die Hersteller auch von Klasse-1-Produkten verpflichtet, Field Safety Corrective Actions (FSCA) zu melden. Solange die EUDAMED nicht in Betrieb ist, gehen diese Meldungen an die Behörden (in Deutschland BfArM, in der Schweiz SwissMedic). Später werden diese Meldungen in der EUDAMED erfasst.
Relativ umfangreich beschreibt das MDCG-Dokument die Pflichten im Fall von FSCA, wie sie zurzeit (noch) die Medizinprodukte-Sicherheitsplanverordnung festlegt.
Schritt 8.c: Umgang mit nicht-konformen Produkten
Der letzte Schritt betrifft den Umgang mit nicht-konformen Produkten. Abgesehen von den bereits erwähnten Meldepflichten (Schritt 8.b) müssen Hersteller entsprechende Korrekturen bzw. korrektive Maßnahmen ergreifen.

Der schnellste und kostengünstigste Weg Ihr Klasse I Produkt zuzulassen
Mit unseren Videotrainings und duzenden Vorlagen erhalten Sie alles, was Sie für eine schnelle und MDR/IVDR-konforme Zulassung brauchen. Ein individuelles Onboarding und ein auf Sie zugeschnittener Projektplan machen es Ihnen leicht, den Überblick zu behalten und alle notwendigen Schritte zu durchlaufen.
3. Was Behörden bei Klasse-I-Produkten prüfen
Anders als bei höherklassigen Produkten obliegt die Überwachung von Klasse-I-Produkten und deren Herstellern v. a. den Behörden. Diese Behörden, z. B. Gewerbeaufsichtsämter und Regierungspräsidien, kommen dieser Überwachungspflicht zunehmend nach. Sie setzen dabei auf Checklisten, wie die folgende beispielhafte Liste. Bitte beachten Sie jedoch, dass nicht alle Checklisten gleich aussehen.
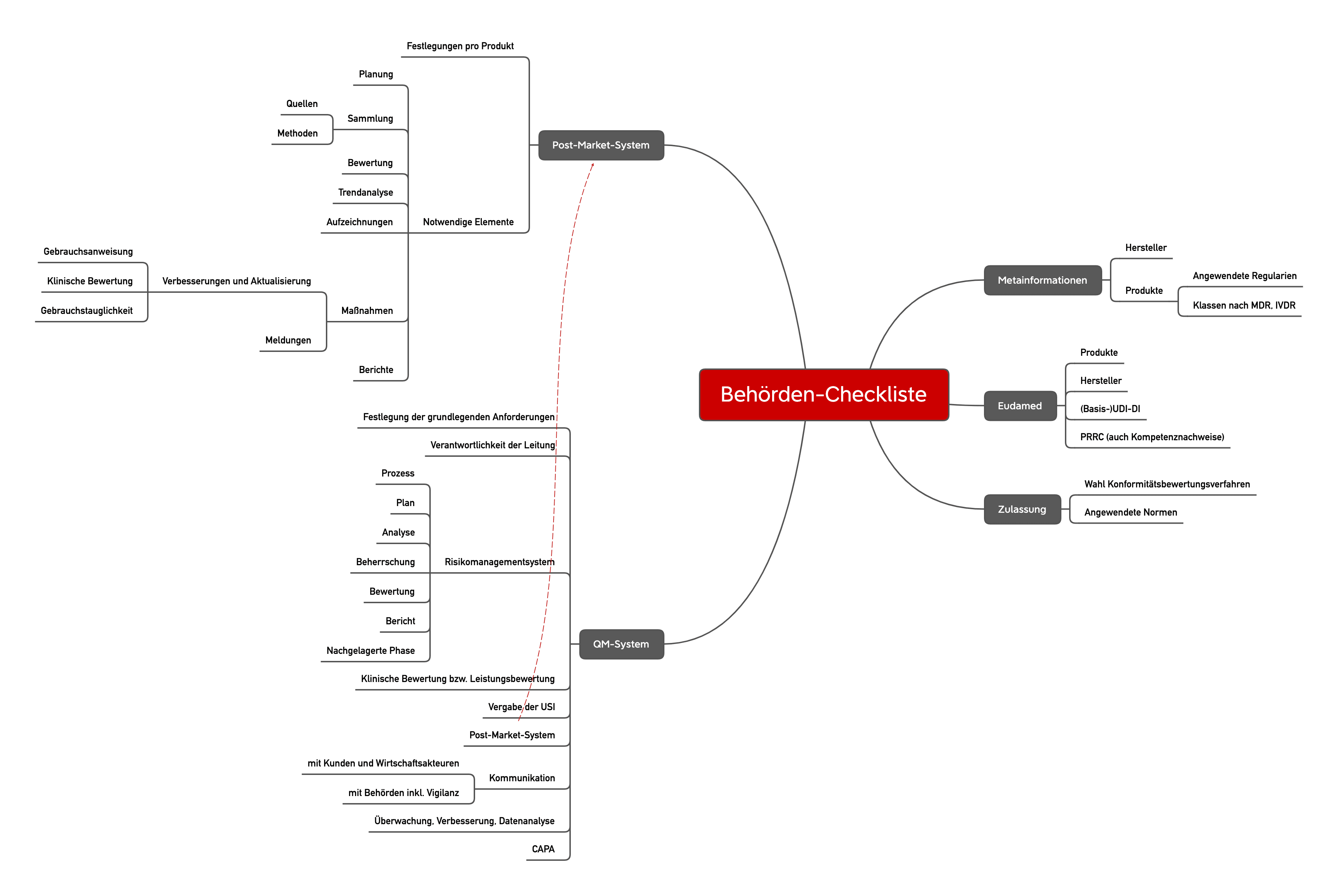
Die Checklisten der Behörden orientieren sich erwartungsgemäß stark an der MDR bzw. der IVDR. Überraschender liegt der Fokus dieser Checkliste auf den Vorgaben zum Risikomanagementsystem, zur Post-Market Surveillance und zur Vigilanz. Hier gehen die Behörden auf fast jeden Teilsatz der regulatorischen Vorgaben ein.
4. Fazit
Das 20-seitige Dokument Guidance Notes for Manufacturers of Class I Medical Devices soll – wie der Name sagt – Herstellern von Medizinprodukten der Klasse I als Leitfaden dienen.
Diesem Ziel wird das Dokument gerecht. Andererseits muss klar sein, dass die darin genannten Anforderungen nicht vollständig und im Wesentlichen nicht spezifisch für Medizinprodukte der Klasse 1 sind.
Das Johner Institut unterstützt Klasse-I-Hersteller dabei, die Konformität mit regulatorischen Anforderungen schnell und einfach zu erfüllen und somit auf Inspektionen durch Behörden perfekt vorbereitet zu sein. Nehmen Sie einfach Kontakt mit uns auf, z. B. über unser Webformular.
Änderungshistorie
- 2025-03-28: Beispiele in Abschnitt 1. a) eingefügt
- 2023-03-04: Hinweise auf Artikel zu Klasse-I-Software ergänzt
- 2021-09-26: Überschriften nummeriert und Kapitel 3 eingefügt.


Sehr geehrter Herr Johner,
herzlichen Dank für die schöne Zusammenfassung des Dokuments. Dazu zwei Fragen:
1) Wo ist dieses Dokument einsehbar? Auf den offiziellen EU-seiten mit MDCG Dokumenten scheint es noch nicht verfügbar zu sein.
2) Die Aussage „Anschließend ist es die Aufgabe der Hersteller, die Medizinprodukte zu registrieren. Dieses Mal bekommen sie eine UDI-DI und Basis-UDI-DI zugeteilt.“ irritiert mich. Zuteilung einer UDI bei der Registrierung in EUDAMED?
Mit besten Grüßen
Mark Hastenteufel
Sehr geehrter Herr Dr. Hastenteufel,
danke für Ihre Fragen!
ad 1) Dass das Dokument noch nicht auf der offiziellen Seite verfügbar ist, ist korrekt. Es ist im Draft-Status.
ad 2) Es benötigt einer Stelle, die die (Basis-)UDI-DIs vergibt bzw. verwaltet. Die Hersteller können nur die PIs verwalten. Der Artikel sagt nicht, dass die EUDAMED diese Verwaltung macht. Vielleicht habe ich das missverständlich formuliert.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich finde auch die Zusammenfassung sehr hilfreich und hätte eine Frage zum Thema UDI:
MDR Art. 120 nennt 3 Stellen: GS1, HIBCC und ICCBBA als benannte Zuteilungsstellen für UDI.
Ist es richtig, dass wir als Hersteller einer dieser Stellen für die Zuteilung einer UDI- DI beauftragen müssen um die UDI vor der Registrierung in EUDAMED zu erhalten?
Unser Medizinprodukt ist ein Software Klasse I nach MDD und Klasse IIa nach MDR.
Beste Grüße
Andrea Miklos
Sehr geehrte Frau Miklos,
es ist wie Sie sagen: Diese „Stellen“ vergeben die UDI-DIs, die Sie für die Registrierung Ihrer Produkte in der Eudamed benötigen.
Welche Klasse das Produkt vor der MDR hat, ist unerheblich. Bei Software gibt es allerdings spezielle Regeln, was die Vergabe der UDI-PIs angeht.
Beste Grüße (auch an DB), Christian Johner
Sehr geehrter Professor Johner,
danke für diesen zusammenfassenden Artikel.
Eine Frage:
Sie schreiben „das „Label“ müsste klarstellen, dass das Produkt ein Medizinprodukt ist“
Gibt es ein Symbol „Medizinprodukt“ oder wie kann diese Vorgabe erfüllt werden und damit klarstellen, dass es keine xyz ist, sondern eben ein Medizinprodukt?
Danke vorab und viele Grüße
Andreas Keller
Sehr geehrter Herr Keller,
noch gibt es das Symbol nicht, weshalb viele einfach „Medizinprodukt“ schreiben. Zusätzlich bedarf es des CE-Zeichens, ab Klasse I* mit Nummer der benannten Stelle.
Beste Grüße, Christina Johner
Sehr geehrter Professor Johner,
in dem neuesten Entwurf der ISO 15223-1 ist unter 5.7.4 ein Symbol definiert, das den Zweck erfüllen kann. Es besteht aus den Großbuchstaben „MD“ und einem Rahmen darum. Bleibt abzuwarten ob es dabei bleibt.
Beste Grüße
Klaus Steger
Danke für den Tipp! Das ist wertvoll!
Danke, lieber Herr Steger!
Beste Grüße, Christian Johner
Guten Tag,
verliert ein Instrument der Klasse 1r seine Zulassung, wenn im Rahmen der Instandsetzung durch einen zertifizierten „Nichthersteller“ die Oberfläche aufbereitet wird (inklusiv Entfernung der vorhandenen Labels mit anschließender Neugravur)? Darf er als Nichthersteller die Zulassungskennung (CE oder MD) anbringen?
Die Produkteigenschaften sowie Zweckbestimmung ändern sich ja nicht. Es ist jetzt schon nicht ganz eindeutig.
Danke.
Herzliche Grüße
Alex Bammert
Sehr geehrter Herr Bammert,
danke für die spannende Frage!
So wie ich das lese, liegt sogar eine Neuaufbereitung vor. Damit wird diese Organisation zum Hersteller:
30. „Hersteller“ bezeichnet eine natürliche oder juristische Person, die ein Produkt herstellt oder als neu aufbereitet bzw. entwickeln, herstellen oder als neu aufbereiten lässt und dieses Produkt unter ihrem eigenen Namen oder ihrer eigenen Marke vermarktet;
Meines Erachtens geht das Eingravieren des CE-Zeichens etwas über das hinaus, was unter Aufbereitung verstanden wird:
39. „Aufbereitung“ bezeichnet ein Verfahren, dem ein gebrauchtes Produkt unterzogen wird, damit es sicher wiederverwendet werden kann; zu diesen Verfahren gehören Reinigung, Desinfektion, Sterilisation und ähnliche Verfahren sowie Prüfungen und Wiederherstellung der technischen und funktionellen Sicherheit des gebrauchten Produkts;
Falls es allerdings keine Neu-Aufbereitung, sondern eine Aufbereitung wäre, ist die Wiederherstellung des CE-Zeichens möglich. Dabei wäre zu klären, in welchem Auftrag diese Aufbereitung erfolgt. Falls dies in Ihrem Namen/Auftrag wäre, würde es sich um einen ausgelagerten Prozess handeln, den Ihr QMS abdecken muss.
Beste Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
eine Frage zum wohl schwierigsten Block, der „Klinischen Bewertung“. Hier für Kl. 1 Produkte.
Ich kann die Verhältnismäßigkeit zwischen den Klassen hierfür nicht erkennen. Wenn z.B. ein Hersteller Mundspatel in Verkehr bringen möchte, sollten doch andere Bedingungen gelten als bei MP Klasse 2 usw.
Eine wirklich gute Klinische Studie verursacht Kosten wohl im 5/6-stelligen Bereich und ist zudem auch ein erheblicher Zeitfaktor. Anderseits ist die Beweisführung / Auswertung über (wissenschaftliche) (Vergleichs-) Literatur ab der MDR wohl leider nicht mehr zulässig?
Freundliche Grüße!
Sehr geehrter Herr Wiedel,
besten Dank für Ihren Kommentar!
Sie formulieren möglicherweise eine Frage, auf die Sie eine Antwort wünschen, nämlich ob die klinische Bewertung (ich mag Ihren Ausdruck der „Beweisführung“) anhand von Literaturdaten und ohne eine klinische Prüfung unter der MDR noch möglich ist.
Die Antwort ist eindeutig, dass dies weiterhin der Fall ist. Im Vergleich zur MDD sind allerdings die Anforderung an die Äquivalenz der Vergleichsprodukte gestiegen. Das hatte streng genommen bereits die MEDDEV 2.7/1 in ihrer vierten Revision vorweggenommen.
Mit den besten Grüßen, Christian Johner
Benötigt man unbedingt eine „benannte Stelle“ wenn man das Könformitätsbewertungsverfahren nach Anhand lX der 2012/745 durchführt? Es geht dabei um Medizinprodukt der Klasse 1.
Sehr geehrter Herr Shchurkov,
wenn Sie die MDR meinen (2017/745), dann kann ich helfen: Für Klasse-I-Produkte benötigen Sie keine benannte Stelle. Für Klasse-I*-Produkte schon.
Beachten Sie, dass die MDR verschoben wurde.
Viele Grüße
Christian Johner
Hallo Herr Dr. Johner,
habe ich es richtig verstanden, dass bei einer Selbstzertifzierung eines Klasse I Produktes ein QM-System vorhanden sein muss, welches allerdings nicht zertifiziert werden muss?
mfg
Gerd Franz
Genauso ist es, Herr Franz!
Wohlgemerkt, Produkte der Klasse I, nicht der Klasse Is, Ir und Im.
Viele Grüße, Christian Johner
Sehr geehrter Prof. Dr. Christian Johner,
Laut MPG § 25 besteht eine allgemeine Anzeigepflicht, trifft das auch auf ein med. Produkt der Klasse I, muss man das Produkt bei DIMDI registrieren?
Das Produkt entspricht nicht der Klasse Is, Ir oder Im.
Die Anzeigepflicht betrifft alle Hersteller, auch die aller Klasse-I-Produkte.
Guten Tag, Danke für die klare Beschreibung! Meine Frage ist noch: Wenn ich einmal die Konformitätserklärung erstellt habe, was mache ich dann damit? Muss ich bei jedem Verkauf meines Medizinproduktes eine Kopie beilegen oder reicht es, wenn ich es aufbewahre für den Fall, dass mich jemand danach fragt und zusätzlich meinen Händlern usw. zur Verfügung stelle?
Sie müssen die Konformitätserklärung nicht mit ausliefern, obwohl das die Hersteller oft tun z.B. in der Gebrauchsanweisung.
Sie müssen aber den Behörden auf Verlangen diese Konformitätserklärung vorlegen.
Sie müssen sie auch den Händlern zur Verfügung stellen, denn diese müssen deren Existenz prüfen.
Guten Tag, ich würde gerne wissen, wie die Dokumentation aussehen muss, wenn man als Hersteller auftritt, das Produkt wird jedoch in einer anderen Firma hergestellt (produziert). Welche Anforderungen und Auflagen kommen auf die Produktionsfirma zu? und wie muss die Arbeit dort in meine Zertifizierung einfließen?
Sehr geehrter Herr Neubarth,
die regulatorischen Anforderungen beziehen sich zuerst auf Sie. Sie müssen die Konformität Ihrer Produkte und Ihres QM-Systems nachweisen. Ein Teil dieser Nachweise werden Sie möglicherweise nicht selbst erbringen können, sondern nur Ihre Produktionsfirma. Beispiele dafür könnten sein:
Ihr QM-System darf keine Lücken aufweisen. Wenn Sie ausgelagerte Prozesse wie die Produktion haben, müssen Sie diese lenken („an die Kandare nehmen“). Wie sehr, das hängt von den Prozessen, von den Risiken und von Ihren Möglichkeiten ab, Abweichungen zu erkennen und zu beseitigen.
Beste Grüße, Christian Johner
Danke Ihnen für die Zusammenfassung. Sie schreiben die Konformitätserklärung ist in jene Sprachen zu übersetzen in denen das Produkt angeboten, sprich verkauft wird. Bedeutet dies im Schluss, wenn das Produkt in Spanien verkauft wird, muss eine Konfi in spanisch zur Verfügung stehen? Wie verhält es sich, wenn Händler in andere Märkte verkaufen. Wer stellt dann die Konfi in der entsprechenden Landessprache?
Mit freundlichen Grüßen
Sehr geehrte Frau Lösche,
danke für Ihre wichtige Frage!
Artikel 19 der MDR fordert:
Damit müsste in Spanien die Konformitätserklärung in spanisch vorliegen. Wenn der Händler in andere Länder verkauft, muss er dafür sorge Tragen, dass diese Erklärung vorliegt (in diesem Fall in der passenden Sprache). Das fordert der Artikel 14, Absatz 2.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für den interessanten Beitrag.
Wir möchten gerne ein Medizinprodukt der Klasse I welches zudem die Zweckbestimmung einer PSA hat von einem Nicht EU-Hersteller mit unserem eigenen Markenname in Deutschland vertreiben.
Somit fallen wir ja in die Importeur-Rolle und haben u.a. die Pflicht die Konfi zu überprüfen, und wie sie schon geschrieben haben müssen die Konfis in Landessprache ausgestellt werden. Dazu habe ich im MPDG folgendes gefunden:
§ 8 Sprachenregelung für die EU-Konformitätserklärung und für Produktinformationen
(1) Der Hersteller hat für Produkte, die im Geltungsbereich dieses Gesetzes auf dem Markt bereitgestellt werden, die
EU-Konformitätserklärung nach Artikel 19 Absatz 1 der Verordnung (EU) 2017/745 in deutscher oder in englischer
Sprache zur Verfügung zu stellen.
(2) Produkte dürfen im Geltungsbereich dieses Gesetzes nur dann an Anwender und Patienten abgegeben
werden, wenn die für Anwender und Patienten bestimmten Informationen in deutscher Sprache zur Verfügung
gestellt werden. In begründeten Fällen dürfen die Informationen auch in englischer Sprache oder einer anderen
für den Anwender des Medizinproduktes leicht verständlichen Sprache zur Verfügung gestellt werden, wenn
diese Informationen ausschließlich für professionelle Anwender bestimmt sind und die sicherheitsbezogenen
Informationen auch in deutscher Sprache oder in der Sprache des Anwenders zur Verfügung gestellt werden
Was heißt das jetzt für uns? Reicht die Konfi in englisch aus?
Vielen Dank im voraus.
Lars Quadt
Sehr geehrter Herr Quadt,
danke für Ihre spannende Frage!
Ich verstehe diese wie folgt: Sie wollen wissen, ob die Konformitätserklärung nur auf Englisch vorliegen kann, auch wenn das MPDG die Informationen für Anwender und Patienten auf deutsch verlangt.
Die kurze Antwort ist ja. Die Konformitätserklärung zählt nicht zu sicherheitsbezogenen Informationen. Daher überschreibt der zweite Absatz nicht den ersten.
Also gute Nachricht: die Konformitätserklärung nur auf Englisch reicht aus.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
uns beschäftigt intern nach wie vor eine Frage, bei der wir keine klare Antwort haben. In Hinblick auf die MDR und die Klasse I Produkte stellt sich uns die Frage, wie es sich mit nicht-sterilen dentalen Einmalprodukten verhält, welche vor der einmaligen Anwendung vom Behandler aufbereitet (gereinigt & sterilisiert) werden müssen. Grundsätzlich handelt es sich ja um Einmalprodukte und nicht um wiederverwendbare Produkte, jedoch mit der eben erwähnten Aufbereitung vor der einmaligen Anwendung. Welcher Klasse sind diese Produkte zugeordnet? Gehören diese noch zur Klasse I oder bereits zur Klasse Ir?
Vielen Dank für eventuell Hinweise oder Gedankenstützen bereits im Voraus!
LG Tobias Bauer
Sehr geehrter Herr Bauer,
danke für Ihre Frage!
Die Klasse Ir bezieht sich auf wiederverwendbare chirurgische Instrumente. Diese Klasse regelt in Artikel 52(7) das dafür zu verwendende Konformitätsbewertungsverfahren.
Die MDR definiert ein „Wiederverwendbares chirurgisches Instrument“ als
Zuerst wäre zu klären, ob Ihre Produkte den Schneiden, Bohren, Sägen usw. dienen. Dann muss man prüfen, ob der Hersteller, das Produkt für die Wiederverwendung durch Reinigung, Desinfektion und Sterilisation bestimmt. So wie ich sie verstehe, schließt das der Hersteller gerade aus. Damit fällt das Produkt nicht in die Klasse.
D.h. wir haben eine Aufbereitung eines Einmalprodukts. Hier greift der Artikel 17. D.h. der Wiederaufbereiter übernimmt Herstellerpflichten. Aber der Hersteller des Produkts wird deshalb nicht zum Hersteller eines Klasse Ir Produkts.
Hilft das als Hinweis?
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich beziehe mich auf den Schritt 7 zu Registrierung von Produkten.
Müssen Klasse I Produkte, die ordnungsgemäß im Medizinprodukte-Informationssystem (MPI) gemäß MDD angezeigt worden sind, die aber unter der MDR weiterhin Klasse I Produkte bleiben, vor/nach Geltungsbeginn der MDR im MPI umgemeldet werden? Muss in einem solchen Fall eine Änderungsanzeige beim DIMDI erfolgen? Oder müssen die Klasse I Produkte unter der MDR beim DIMDI neu registriert werden?
Vielen Dank im Voraus!
Freundliche Grüße
Ilona Milke
Sehr geehrte Frau Milke,
danke für Ihre wichtige Frage!
Derzeit können Sie gar nichts tun, weil die EUDAMED noch nicht soweit ist. D.h. es bleibt vorläufig bei der Pflicht sich im deutschen Informationssystem zu registrieren.
Das DIMDI gibt es nicht mehr. Es wurde vom BfArM „übernommen“. Das Informationssystem bleibt aber das gleiche.
Medizinprodukte, die in der Klasse I bleiben, müssen Sie somit nur dann neu anmelden/ummelden, wenn sich etwas ändert, z.B. wenn Sie die Konformitätserklärung bezüglich der MDR statt der MDD ausstellen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
chirurgische Instrumente fielen unter der MDD in die Klasse I und bedurften nicht der Einbindung einer benannten Stelle. Mit der MDR 2017/745 fallen diese in die Klasse I r die nun diese Einbindung erforderlich macht. Im Workshop der DGRA Bonn (Unter Teilnahme von Führungskräften des BGM, BfArM und EU-Rechtlern) wurde erklärt, dass es für die neue Klasse I r keine Übergangsfristen wie für alle anderen Klassen gäbe. Mich hat stutzig gemacht dass Mitbewerber im Sektor minimal-invasiv endoskopische Wirbelsäulenchirurgie über kein Zertifikat nach MDR 2017/745 verfügen, ihre Produkte aber trotzdem Europa- und Weltweit verkaufen. Bei meiner Recherche bin ich auf das Dokument „Corrigendum II“ der EU gestoßen, indem festgestellt wird, dass der entsprechende Artikel geändert wurde und für Medizinprodukte Klasse I r nun doch eine Übergangsfrist bis 2024 gilt. Das würde bedeuten, dass eine Einbindung von benannten Stellen erst ab diesem Zeitpunkt erfolgt sein muss. Das wäre nur logisch, da die Etablierung von benannten Stellen nicht dem Bedarf folgt. Bitte senden Sie mir eine Stellungnahme aus Ihrer Sicht.
Mit besten Grüßen
Friedrich Tieber
Sehr geehrter Herr Tieber,
für Klasse Ir Produkte gibt es eine Übergangsfrist. Details können Sie dem Artikel 120 sowie unseren Ablaufdiagramm am Endes des Artikels zu den Übergangsfristen entnehmen.
Dass etwas anderes behauptet wird, erstaunt mich. Haben Sie zufällig Unterlagen? Möglicherweise gibt es ein Missverständnis, das ich helfen kann aufzulösen. Falls Sie etwas hätten, dann schicken Sie es gerne an [email protected].
Viele Grüße, Christian Johner
Sehr geehrte Damen und Herren,
medizinsiche Einwegmasken (wenn man Sie so denn überhaupt bezeichnen darf, viele benutzen „medizinscher Mund NasenSchutz“) gehören zu Medizinprodukt der Klasse 1. Der Hersteller (in China) hat einen EC Rep , die Masken wurde durch den EC Rep nach §25 und §30 MPG angezeigt.
Ein deutsches Unternehmen bezieht beim Hersteller in China diese Masken und importiert diese. Das deutsche Unternehmen hat vorher keine Medizinprodukte importiert.
1. Muss der Import dieser Masken angezeigt werden
2. Unterliegt das deutsche Unternehmen Auflagen in Sachen QM, Dokumentation, Sicherheitsbeauftragten, etc. ?
Sehr geehrter Herr Kiel,
Unternehmen, die Medizinprodukte importierten, müssen die Anforderungen an Importeure gemäß MDR bzw. IVDR erfüllen. Beachten Sie dazu den Artikel über Importeure.
Bevor das Produkt aber verkauf werden kann, muss es ein Konformitätsbewertungsverfahren durchlaufen haben, je nach Produkt unter Einbeziehung einer Benannten Stelle.
Wenn der Hersteller des Produkts außerhalb der EU seinen Sitz hat, dann bedarf es zusätzlich eine EU-Bevollmächtigten.
In jedem Fall müssen alle Medizinprodukte registriert sein, ebenso die Hersteller und andere Wirtschaftsakteure.
Beachten Sie, dass MPG durch das MPDG abgelöst wurde.
Viele Grüße, Christian Johner
Hallo,
verstehe ich das richtig, dass ein Risikomanagement nach ISO 14971 bei Klasse 1 ohne Zusatz somit nicht nötig ist? Es ist ausreichend Risko-Nutzen im Rahmen der Klinischen Bewertung zu erschlagen?
Beste Grüße
Ben Mohr
Sehr geehrter Herr Mohr,
die Anforderungen an das Risikomanagementsystem hängen nicht(!) von der Klassifizierung ab. Sie brauchen somit immer eine vollständige Risikomanagementakte, die man üblicherweise konform den Vorgaben der ISO 14971 erstellt.
Das Risiko-Nutzen-Verhältnis nur in der klinischen Bewertung zu diskutieren, reicht somit nicht aus.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner, im zusammenhang das ein Medizinprodukt der Klasse 1 keine benante Stelle bewertet wird ist ok Die Frage ,die sich stellt , wenn ein Medizinprodukt der Klasse 3 mit einem Operationsprodukt in einer sterilen Box verpackt wird kann es dann als „Nichtmedizinprodukt “
deklariert werden???
Sehr geehrte Frau Banasiewicz,
wenn sich die Zweckbestimmung des Medizinproduktes nicht geändert hat, sehe ich keine Möglichkeit, die Zusammenstellung als „Nicht-Medizinprodukt“ anzusehen. Des Weiteren würde ich vermuten, dass Sie ein System oder gar eine Behandlungseinheit erzeugt haben. Weiteres zu Systemen / Behandlungseinheiten können Sie unter folgendem Link einsehen.
Herzliche Grüße
Guten Tag,
vielen Dank für die hervorragend aufbereiteten Unterlagen. Sie erleichtern mir den Einstieg in das Thema sehr!
Leider hat sich der Fehlerteufel eingeschlichen:
Field Saftey Correction Actions kürzen sich mit FSCA ab, nicht mit FCSA.
Viele Grüße
Dr. Silke Griemsmann
Sehr geehrte Frau Dr. Griemsmann,
vielen Dank für diesen wertvollen Hinweis. Ich habe diesen Fehler soeben korrigiert.
Nochmals herzlichen Dank für Ihren Hinweis.
Herzliche Grüße
Guten Tag,
Leider konnte ich die Anforderungen für meine Medizinprodukte der Klasse II weder in diesem Beitrag noch in einem anderen finden. Ich würde mich auf Ihre Hilfe freuen.
Mit freundlichen Grüßen,
Aida Erme
Sehr geehrte Frau Erme,
wir haben in der Tat keinen eigenen Beitrag für die Medizinprodukte der Klassen IIa und IIb. Für Sie wichtig zu wissen ist, dass
Wir haben sehr viele Beiträge auch für diese Produkte. Mein Tipp wäre, dass Sie mit diesem Beitrag zum Einstieg beginnen.
Geben Sie gerne Bescheid, wenn wir Sie weiter unterstützen können.
Beste Grüße, Christian Johner
Guten Tag Herr Professor Johner,
wir sind ein Start-Up und wollen eine innovative Pillenbox mit 4*7 Fächern launchen.
Von potentiellen Abnehmern haben wir jetzt gehört, dass die Pillenbox als Medizinprodukt der Klasse 1 zugelassen werden muss. Das würde erhebliche Kosten nach sich ziehen.
Aus den Gesetzestexten kann ich die Notwendigkeit auch nicht ableiten, da die Pillenbox nicht unmittelbar dafür bestimmt ist, folgenden medizinischen Zweck zu erfüllen: Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten.
Oder sehe ich das zu naiv?
Vielen Dank für Ihre Hilfe!
Georg Hachmann
Sehr geehrter Herr Hachmann,
danke für Ihre spannende Frage!
Um eine Antwort darauf zu geben, müsste ich die Zweckbestimmung kennen. Denn „Pillenbox mit 4*7 Fächern“ ist eher eine Funktionsbeschreibung. Entscheidend ist aber die von Ihnen definierte Zweckbestimmung.
Bei gleicher Funktionalität kann ein identisches Produkt bei unterschiedlicher Zweckbestimmung einmal als MP und einmal als Nicht-MP qualifizieren.
Wir unterstützen Sie dabei, die Zweckbestimmung so zu formulieren, dass sie wasserdicht ist und Sie nicht in Gefahr laufen, verklagt zu werden. Denn dann wird es oft schwieriger zu helfen. Schreiben Sie uns einfach oder nutzen Sie das Kontaktformular oben rechts.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner!
Zuerst einmal ein herzliches Danke für diese wunderbare und frei Verfügbare Fundgrube und Wissendatenbank!
Zum Thema:
Ich kann nicht ganz nachvollziehen, wie Sie im Text unter 1.b Klasse-1 Produkte mit dem Satz „Zudem besteht die MDR bei Medizinprodukten der Klasse 1 nicht auf der Zertifizierung des Qualitätsmanagementsystems durch eine Benannte Stelle.“ von der Verpflichtung der Bewertung des QM-Systems durch die benannte Stelle lt. Annex IX generell ausnehmen können.
Denn ich lese den Artikel 52 so, dass vor jedem In-Verkehrbringen wie auch vor jeder In-Betriebname eines noch nicht In-Verkehrgebrachten Medizinproduktes auch Klasse I Produkte betroffen sind (Artikel 52 (1) & (2)) und somit die Anwendung des kompletten Annex IX, Annex X oder XI vorgeschrieben wird und man für die In-Verkehrbringung die „EU-Qualitätsmanagementbescheinigung“ halten muss.
Die (7) mit dessen Reduktion auf die Erstellung des TechDoc gilt meines Erachtens explizit nur die Herstellung von Medinzinprodukte der Klasse I.
Lese ich falsch, habe ich irgendwelche Ausnahmeregelungen übersehen? Wenn nicht: wie wird das Gesetz diesbezüglich im Feld heutzutage interpretiert?
Sehr geehrter Herr Stift,
ich kann nicht nachvollziehen, wie Sie zu der Annahmen kommen, das Artikel 52 (7) lediglich die „Herstellung“ von Klasse I Medizinprodukten adressiert. Der Begriff „Herstellung“ wird lediglich unter Artikel 52 (7) a) verwendet und hat hierbei keinen grundsätzlichen Charakter.
Viele Grüße
Ich komme zu diesen Schluss, weil Absatz 7 mit den Worten „Hersteller von Produkten der Klasse I“ und nicht mit „In-Verkehrbringer von Produkten der Klasse I“ beginnt, gefolgt mit den reduzierten Anforderung und einer Reihe an Ausnahmen von dieser Reduktion.
Es geht also eigentlich nicht um Absatz 7, sondern darum, dass in Absatz (1) und (2) keine Klasse ausgenommen wird. Somit gilt für mich, sobald ich In-Verkehrbringer bin, dass ich immer eine „EU-Qualitätsmanagementbescheinigung“ benötige. Also stimmt meines Erachtens die Aussage „Zudem besteht die MDR bei Medizinprodukten der Klasse 1 nicht auf der Zertifizierung des Qualitätsmanagementsystems durch eine Benannte Stelle.“ nicht (außer ich habe im Gesetz die explizite Ausnahme von Klasse I überlesen), sondern trifft nur auf den Fall zu, wenn man Hersteller von einem schon auf dem EU-Markt eingeführten Produkt ist.
Hier die Absätze aus Artikel 52 der MDR:
In Absatz (1) kann ich keine Einschränkung auf eine Klasse finden:
„(1) Bevor Hersteller ein Produkt in Verkehr bringen, führen sie eine Bewertung der Konformität des betreffenden Produkts im Einklang mit den in den Anhängen IX bis XI aufgeführten geltenden Konformitätsbewertungsverfahren durch.“
„(7) Hersteller von Produkten der Klasse I, ausgenommen Sonderanfertigungen oder Prüfprodukte, erklären die Konformität ihrer Produkte durch Ausstellung einer EU-Konformitätserklärung gemäß Artikel 19, nachdem sie die technische Dokumentation gemäß den Anhängen II und III erstellt haben. Bei Produkten, die in sterilem Zustand in den Verkehr gebracht werden, bei Produkten mit Messfunktion oder bei Produkten, bei denen es sich um wiederverwendbare chirurgische Instrumente handelt, wendet der Hersteller die in Anhang IX Kapitel I und III oder in Anhang XI Teil A aufgeführten Verfahren an. Die Beteiligung der Benannten Stelle an diesen Verfahren ist jedoch begrenzt
a) bei Produkten, die in sterilem Zustand in Verkehr gebracht werden, auf die Aspekte, die mit der Herstellung, der
Sicherung und der Aufrechterhaltung steriler Bedingungen zusammenhängen,
b) bei Produkten mit Messfunktion auf die Aspekte, die mit der Konformität der Produkte mit den messtechnischen
Anforderungen zusammenhängen,
c) bei wiederverwendbaren chirurgischen Instrumenten auf die Aspekte, die mit der Wiederverwendung in
Zusammenhang stehen, insbesondere die Reinigung, Desinfektion, Sterilisation, Wartung und Funktionsprüfung
sowie die damit verbundenen Gebrauchsanweisungen.“
Somit gilt für mich, wenn ich In-Verkehrbringer bin, dann muss ich zum Zeitpunkt der Einführung eine Bewertung meines Management System durch die benannte Stelle beantragt haben:
Annex IX, Kapitel I, Absatz 2. Bewertung des Qualitätsmanagementsystems
„2.1. Der Hersteller beantragt bei einer Benannten Stelle die Bewertung seines Qualitätsmanagementsystems. …“
Zumindest die Wiederbewertung des Mgmt-Systems fällt laut Absatz 3 für Klasse I Produkte weg:
„3. Überwachungsbewertung bei Produkten der Klasse IIa, der Klasse IIb und der Klasse III“
Ich hoffe, dass ich es jetzt verständlicher geschrieben habe.
Mit freundlichen Grüßen und vielen Dank für ihre Mühen!
Sehr geehrter Herr Stift,
vielen Dank für die weitere Ausführung der zugrunde liegenden Fragestellung. Leider folgt die MDR keiner mir bekannten logischen Struktur. Dies zeigt sich auch darin, dass Begrifflichkeiten unterschiedlich oder unpräzise verwendet werden. Bzgl. Ihrer Fragestellung/Anmerkung sind wir der Auffassung, dass sich Unterpunkt 1 von Artikel 52 auf Produkte bezieht, welche den Einbezug einer Benannten Stelle vorsehen aufgrund der aufgeführten Konformitätsbewertungsverfahren mit Anhang IX bis XI. Unterpunkt 7 von Artikel 52 adressiert Produkte der Klasse I, für welche keine Benannte Stelle benötigt werden.
Diese Sichtweise wird unserer Erfahrung nach auch durch die Handhabung im Markt seitens der Aufsichtsbehörden derart gelebt. Wichtig ist hierbei zu erwähnen, dass sehr wohl ein zertifiziertes Qualitätsmanagementsystem (seitens Zertifizierstelle) gewünscht ist.
Herzliche Grüße
Sehr geehrter Herr Prof. Dr. Johner,
in unserer Firma diskutieren wir immer noch über die Klassifizierung des Medizinprodukts, bei der wir bisher noch keine klare Antwort bzw. Begründung haben. Bezüglich der Klasse 1-Produkte stellen sich uns einige Fragen, wie es sich mit steril gelieferten Tropfer zu unseren Kunden, welche zum Einbau in ein Endprodukt als auch zur weiteren Verarbeitung, um das Endprodukt zu vermarkten.
Es geht hierbei um Tropfer / Pumpe, die zur Verabreichung der Flüssigkeit (Arzneimittel) dienen. Wir als Hersteller der Pumpe liefern unsere Produkte an unseren Kunden im sterilen Zustand aus. Diese werden von unseren Kunden weiterverarbeitet und aseptisch befüllt. Daher wird das finale Produkt (als Augen- / Nasentropfen), welches eine andere Risikoklasse auf dem Markt verkauft wird. Für das finale Produkt trägt die Verantwortung als Hersteller unsere Kunden. Wir möchten dann die Herstellerpflicht ausschließlich für die Pumpe übernehmen. Wir nehmen an, dass unsere Pumpe als System (Art. 2 (11)) definiert wird, welches zur späteren Weiterverarbeitung kombiniert werden soll. Die Fragen, die ich habe, beziehen sich auf die Risikoklasse. Gemäß Info von einem unserer Kunden hat seine Benannte Stelle im Rahmen seiner Einreichung der Stellungnahme Benannte Stelle (NBOp) die Pumpe als Device Part unter Risikoklasse 1 anstatt 1s bewertet, mit der Begründung dass die Pumpe vom Endkunden angefasst werden kann und es daher nicht steril ist.
Allerdings gemäß Artikel 22 (3): „Jede natürliche oder juristische Person, die Systeme oder Behandlungseinheiten gemäß Absatz 1 für ihr Inverkehrbringen sterilisiert, wendet wahlweise eines der Verfahren gemäß Anhang IX oder das Verfahren gemäß Anhang XI Teil A an. Die Anwendung dieser Verfahren und die Beteiligung der Benannten Stelle sind auf die Aspekte des Sterilisationsverfahrens beschränkt, die der Gewährleistung der Sterilität des Produkts bis zur Öffnung oder Beschädigung der Verpackung dienen. Die natürliche oder juristische Person gibt eine Erklärung ab, aus der hervorgeht, dass die Sterilisation gemäß den Anweisungen des Herstellers erfolgt ist.“
Da wir die Pumpe zum Kunden steril ausliefern aber beim Kunden weiterverarbeitet und dadurch ihre Sterilität verliert, unter welcher Klasse ist die Pumpe zugeordnet? Wird sie noch zur Klasse 1 mit der obigen Begründung gehören oder besser Klasse 1s aufgrund dem Lieferzustand zu unseren Kunden?
Vielen Dank im Voraus für Ihre Antwort.
Viele Grüße
P. Panigoro
Sehr geehrter Herr Panigoro,
wenn sie die Pumpe steril ausliefern und das Produkt auch als „steril“ Kennzeichnen, dann kommen sie kaum um die Klasse Is herum. Hierbei stellt sich mir die Frage, weshalb sie die Pumpe steril ausliefern, wenn diese ohnehin beim Kunden erneut sterilisiert wird. Wenn sie die Pumpe sterilisieren, dies jedoch nicht auf dem Label angeben, also nicht kennzeichnen, dann wäre die Klasse I korrekt.
Herzliche Grüße
Sehr geehrter Gerhart,
erwähnen sollte man vielleicht noch:
Eine Frage die immer wieder auftaucht und zur Verunsicherung bei Händlern führt ist: Bleiben Zahn-Matrizen aus Edelstahl die vom Zahnarzt mehrfach verwendet und sterilisiert werden können weiterhin Klasse I Produkte (nicht Klasse Ir) ? Bekanntermaßen Schneiden, Schaben und Bohren diese Produkte ja nicht (kein chrirurg. Instrument). Oder ändert sich daran etwas? Die Zahn-Matrizen werden seit Jahrzehnten im nicht sterilen Zustand an den Zahnarzt und Händler geliefert.
Herzliche Grüße
Klaus Niemers
Sehr geehrter Herr Niemers,
konnte mein Kollege Mario Klessascheck Ihre Frage bereits beantworten? Falls nicht, dann melden Sie sich gerne nochmal, z. B. über eine kostenlose Microconsulting-Anfrage: https://www.johner-institut.de/micro-consulting/
Herzliche Grüße
Tea Bodrusic
Sehr geehrte Frau Bodrusic,
vielen Dank für Ihre Nachricht.
Leider wurde diese einfache Frage bis jetzt nicht beantwortet.
Herzliche Grüße
Klaus Niemers
Sehr geehrter Herr Niemers,
mir ist nicht bekannt, dass die Klassifizierung von Zahn-Matrizen angepasst wurde oder werden soll. Somit werden diese nach meinem Kenntnisstand weiterhin der Klasse I zugeordnet.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
herzlichen Dank für Ihre eindeutige, aussagekräftige Antwort.
Ich wünsche Ihnen ein schönes Wochenende.
Herzliche Grüße
Klaus Niemers
Sehr geehrter Herr Gerhart,
unsere handbedienten Kugelhähne werden zur Durchleitung von Flüssigkeiten u.a. auch in Medizinprodukten eingesetzt, die in Krankenhäusern verwendet werden. Es fließt durch unsere Kugelhähne kein Medium, das mit dem Patienten in Berührung kommt oder kam. Handelt es sich bei unseren Kugelhähnen nun um ein Medizinprodukt Kl. I? Oder fallen solche „einfachen“ Bauteile/Komponenten nicht unter die MDR?
Viele Grüße
Marc Ottensmeyer
Sehr geehrter Herr Ottensmeyer,
danke für Ihre spannende Frage! Die Entscheidung, ob es sich überhaupt um ein Medizinprodukt handelt, lässt sich kaum treffen, ohne die Zweckbestimmung zu kennen. Beispielsweise ist es hilfreich zu wissen, ob das Produkt speziell für medizinische Zwecke genutzt werden soll oder auch in anderen Kontexten zum Einsatz kommt. Es könnte auch ein Zubehör sein. Dann würden die Anforderungen der MDR gelten.
Nutzen Sie bei weiteren Fragen gerne auch das kostenlose Micro-Consulting, über das wir eine Zweckbestimmung austauschen können.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
welche Normen gelten für Medizinprodukte der Klasse I Produkte (also nicht „1s, 1r und 1m“) die man ja auch auflisten muß in der technischen Dokumentation.
Sind es die nachfolgenden und wo findet man die Aktualisierungen? Habe lange gesucht leider nichts dazu gefunden:
DIN EN 980:2003, Graphische Symbole zur Kennzeichnung von Medi-zinprodukten
ISO 9001:2008, Qualitätsmanagementsysteme, Anforderungen
DIN EN ISO 13485:2010, Medizinprodukte, Qualitätsmanagementsys-teme, Anforderungen für regulatorische Zwecke
ISO 14971:2007, Medizinprodukte – Anwendung des Risikomanage-ments auf Medizinprodukte
Medizinproduktegesetz der Bundesrepublik Deutschland (MPG) v. 14.6.2007
RICHTLINIE 93/42/EWG DES RATES vom 14. Juni 1993 über Medizin-produkte, Medical Device Directive (MDD), letzte Änderung v. 5.9.2007 (RL 2007/47/EG)
Medizinprodukte-Verordnung–MPV vom 20.12.2001, zuletzt geändert am 16.2.2007.
DIMDI-Verordnung-DIMDIV Verordnung über das datenbankgestützte Informationssystem über Medizinprodukte des Deutschen Instituts für Medizinische Dokumentation und Information vom 04. Dezember 2002 (BGBL. I S. 4456)
Medizinprodukte-Sicherheitsplanverordnung-MPSV Verordnung über die Erfassung, Bewertung und Abwehr von Risiken bei Medizinpro-dukten.
Sehr geehrter Herr Niemers,
eine derartige Liste ist mir nicht bekannt und kann es nach meiner Ansicht nur mit Einschränkungen geben, weil diverse Normen nur produktabhängig Anwendung finden. Eine erste Orientierung gibt Ihnen die Liste der harmonisierten Normen der EU, welche Sie unter dem nachfolgenden Link (https://ec.europa.eu/docsroom/documents/55101) finden. Sollten Sie bei der Recherche der produktspezifischen Normen benötigen, zögern Sie nicht, uns zu kontaktieren. Wir helfen sehr gerne.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
herzlichen Dank für Ihre schnelle Antwort.
Ich habe mich vielleicht falsch ausgedrückt. In der „Struktur Technische Dokumentation (Medizinprodukte)“ von mdc steht unter ist unter:
4.2. Liste der angewandten Normen, Gemeinsamen Spezifikationen
Aktuelle Liste der angewandten Normen mit Ausgabestand, sowie gegebenenfalls die Angabe, welche Teile der Normen nicht angewandt wurden. (Dieser Punkt ist gemäß MDR Bestandteil von 4.1, jedoch für Verfahren nach Richtlinie 93/42/EWG explizit erforderlich).
Die Liste muß man offensichtlich aufführen in der Technischen Dokumentaion, nur woher bekommt man diese Liste. Oder fällt dies bei Klasse I (also nicht „1s, 1r und 1m“) weg?
Herzlichen Dank für Ihre Hilfe
Sehr geehrter Herr Niemers,
die Liste der angewandten Normen, Gemeinsamen Spezifikationen ist Bestandteil der technischen Dokumentation und muss unabhängig von der Produktklasse erstellt werden bzw. vorhanden sein. Die Liste muss produktspezifisch erarbeitet/erstellt werden. Als Orientierung können Sie hierzu die Liste der harmonisierten Normen der EU nutzen (siehe vorheriger Kommentar). Diese ist jedoch nicht vollumfänglich. Die Weiteren sinnvoll anzuwendenden Normen, um die Konformität mit den regulatorischen Anforderungen darzulegen, müssen recherchiert und angewendet werden. Gerne können wir Sie hierbei unterstützen.
Herzliche Grüße
Sehr geehrter Herr Gerhart,
vielen Dank für Ihre Antwort. Ich weiß nur nicht wie ich herausbekomme welche Normen zutreffen und welche aufgeführt werden müssen.
Ich habe bereits Ihre erwähnte Liste über den Link zu den harmonisierten Normen durchgesehen.
Hilfreich wäre hier wie man genau vorgehen soll.
Ich weiß nicht wie das herauszufinden ist.
Wie geht das?
Herzliche Grüße
Sehr geehrte/e „Niemers“,
wir sind noch nicht ganz sicher, ob wir Ihre Frage verstehen. Daher hier der Versuch einer Antwort:
Welche Normen zutreffen, hängt von der Art Ihres Medizinprodukts ab. Es gibt wenige Normen, die bei allen Produkten Anwendung finden. Dazu zählen die Normen zum Risikomanagement, zum Usability Engineering und zum Qualitätsmanagement.
Bei Produkten, die in physischen Kontakt mit den Patienten kommen, sind die Normen der Reihe ISO 10993 relevant, bei Produkten, die Strom nutzen, sollten Sie die Normenreihe IEC 60601 in Betracht ziehen.
Mehr können wir ohne genaue Kenntnis Ihres Produkts nicht sagen. Die genaue Liste der regulatorischen Anforderungen und der anwendbaren Normen zu erstellen, ist ein Teil der regulatorischen Strategie. Dabei können wir Sie unterstützen. Nehmen Sie gerne Kontakt auf, damit Ihnen unser Beratungsteam ein maßgeschneidertes Angebot erstellen kann.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Antwort.
Es geht wie weiter oben beschrieben um Klasse I (also nicht „1s, 1r und 1m) so z. B. Pinzetten, Spannzangen etc. Die im Mund angewendet werden.
Vielen Dank.
Beste Grüße
Klaus Niemers
Sehr geehrter Herr Professor Johner,
unsere Firma hat bereits ein nach der DIN EN ISO 13485 zertifiziertes QM-System, ist aber bisher nicht als Legal Supplier aufgetreten.
Wir planen aber zukünftig als Legal Supplier für ein Zubehör zu einem Medizinprodukt der Klasse I (nicht 1m, 1s oder 1r) aufzutreten.
Wenn wir den obigen Artikel richtig verstanden haben, ist es vor der Inverkehrbringung nicht notwendig unser QM-System diesbezüglich vorab zertifizieren zu lassen.
Falls Sie zusätzliche Informationen benötigen, melden Sie sich bitte bei uns.
Wir würden uns auch freuen, wenn Ihre Firma uns als externen Berater betreuen könnte.
Sehr geehrte Frau Schulz,
danke für Ihre Frage!
Sehr gerne helfe ich, bin aber noch nicht sicher, ob ich alles richtig verstanden habe. Es gibt die Rolle des Supliers und die Rolle des Legal Manufacturers. Ich antworte in der Annahme, dass es um letztere Rolle geht:
Ihre Annahme ist korrekt. Sie benötigen ein QM-System, das die Anforderungen der MDR erfüllt. Es ist hilfreich, sich dabei von der ISO 13485 leiten zu lassen, weil diese etwas konkretere Vorgaben macht und auch den Stand der Technik repräsentiert. Aber eine Zertifizierung durch einen Zertifizierer oder gar eine Benannte Stelle ist nicht vorgeschrieben.
Hilft das? Falls nicht haken Sie einfach nach.
Mit besten Grüßen, Christian Johner
Lieber Herr Gerhardt,
Danke für Ihren spannenden Artikel. Benötigt ein Medizinprodukt der Klasse 1 eine EMDN Nummer?
Liebe Frau Henze,
vielen Dank für Ihre Frage. Die kurze und schlichte Antwort ist ja. Sie brauchen die EMDN Nummer zum einen für die Meldung von Zwischenfällen beim BfArM und zum anderen um Meldungen und die Registrierung in der EUDAMED durchführen zu können (siehe hierzu den Fachartikel „EMDN, UMDNS, MDA und weitere Kodiersysteme„).
Herzliche Grüße
Markus Gerhart
Sehr geehrte Damen und Herren,
eine kurze Frage von mir: muss die Rückverfolgbarkeit von Medizinprodukten der Klasse 1 chargenpflichtig bis zum Endkunden gewährleistet sein?
Vielen Dank bereits für die Antwort.
Viele Grüße
Sehr geehrte Frau Kroll,
für Medizinprodukte der Klasse 1 gelten hinsichtlich der Rückverfolgbarkeit keine Ausnahmeregelungen. Somit muss die Charge bis zum Endkunden identifizierbar sein. Genaueres können Sie dem Dokument „Leitfaden für die Umsetzung der Produktvorschriften der EU 2022 (Blue Guide)“ in Kapitel 4.2.2 entnehmen. Sollten Sie hierbei Unterstützung benötigen, können Sie sich sehr gerne direkt an uns wenden, wir helfen sehr gerne.
Herzliche Grüße
Markus Gerhart
Sehr geehrter Herr Gerhart,
vielen Dank für die Antwort.
Herzliche Grüße Franka Kroll
Sehr geehrter Herr Gerhart,
Ich habe eine Frage ich plane, ein Unternehmen für chirurgische Instrumente in Deutschland zu gründen, wobei die Herstellung in Pakistan erfolgen soll. Ich möchte chirurgische Instrumente in den Bereichen Allgemeinchirurgie, Zahnmedizin, Schönheitspflege und Tiermedizin vertreiben.
Könnten Sie mir bitte mitteilen, welche Lizenzen und Zertifizierungen ich für den Verkauf dieser Produkte in Deutschland benötige? Insbesondere interessiert mich:
• Die CE-Zertifizierung und Anforderungen gemäß der Medical Device Regulation (MDR)
• Notwendige Registrierungen bei den Behörden
• Mögliche Kosten für die erforderlichen Zertifizierungen und Lizenzen
Wenn es möglich ist, wäre ich auch für weitere Informationen und Unterstützung bei diesem Vorhaben sehr dankbar. Ich freue mich auf Ihre Rückmeldung und wäre dankbar für jegliche Hilfe, die Sie mir anbieten können.
Mit freundlichen Grüßen,
Muhammad Shoaib
Sehr geehrter Herr Shoaib,
vielen Dank für Ihre ausgesprochen umfangreichen Fragen, welche ich leider aufgrund der Komplexität und des Umfangs der notwendigen Antwort an dieser Stelle nicht beantworten kann. Ich habe mir jedoch erlaubt, Ihre Frage an das Beratungsteam weiterzuleiten, welche sich mit Ihnen in Verbindung setzen werden.
Herzliche Grüße
Markus Gerhart