
Die Medizinprodukte-Betreiberverordnung (MPBetreibV) ist eine nationale deutsche Verordnung, die Anforderungen an das Betreiben und Benutzen von Medizinprodukten stellt.
| Vollständige Bezeichnung | Verordnung über das Betreiben und Benutzen von Medizinprodukten |
|---|---|
| Kurztitel | Medizinprodukte-Betreiberverordnung |
| Abkürzung | MPBetreibV |
| Gesetzestext | MPBetreibV als kompletter Text |
1. FAQ zur MPBetreibV
a) Für wen gilt die MPBetreibV?
Die Medizinprodukte-Betreiberverordnung gilt für Organisationen und Personen, die Medizinprodukte und In-vitro-Diagnostika betreiben und benutzen.
Typische Organisationen sind Krankenhäuser, Kliniken, Arzt-Praxen, Reha-Einrichtungen und medizinische Labore.
Organisationen und Personen, die Medizinprodukte errichten, bereithalten, instand halten, aufbereiten oder sicherheits- und messtechnischen Kontrollen unterziehen, fallen ebenfalls in den Anwendungsbereich der Verordnung. Mehr dazu in Kapitel 3 dieses Artikels.
Ebenso gilt die MPBetreibV für Versorgende und Dritte, die einem Patienten Produkte zur Benutzung bereitstellen. Bei den Versorgenden handelt es sich z. B. um Krankenkassen, Unfallkassen, Pflegekassen sowie private Krankenversicherungsunternehmen. Dritte sind hingegen bspw. Sanitätshäuser und Apotheken.
Lesen Sie hier mehr über die Pflichten von Gesundheitseinrichtungen (Betreibern).
b) Für welche Produkte gilt die MPBetreibV?
Der Anwendungsbereich der MPBetreibV erstreckt sich gemäß § 1 (1) der Verordnung auf Produkte nach § 3 Nr. 1 des MPDG.
Medizinprodukte, ihr Zubehör und die in Anhang XVI der Verordnung (EU) 2017/745 aufgeführten und unter den Anwendungsbereich dieser Verordnung fallenden Produkte sowie In-vitro-Diagnostika und ihr Zubehör;
Die Vorschriften der MPBetreibV sind jedoch erst ab August 2025 für Produkte aus Anhang XVI der Verordnung (EU) 2017/745 (MDR) anzuwenden.
c) Welches Ziel verfolgt die MPBetreibV?
Ausgestaltung des Gesetzes
Formal verfolgt der Gesetzgeber mit der MPBetreibV das Ziel, die gesetzlichen Anforderungen des Medizinprodukterecht-Durchführungsgesetzes MPDG noch genauer zu formulieren.
Beispielsweise schreibt das MPDG:
Produkte […] dürfen nur nach Maßgabe der Rechtsverordnung nach § 88 Absatz 1 Satz 1 Nummer 6 betrieben und angewendet werden.
§ 11 MPDG
Was beim Betrieb dieser Produkte beachtet werden muss, sagt das MPDG nicht, sondern verweist auf die Rechtsverordnung – die Medizinprodukte-Betreiberverordnung.
Sicherheit von Patienten
Als Teil des Rechtssystems bestehend aus EU-Verordnungen (MDR, IVDR), Gesetzen (MPDG) und anderen nationalen Verordnungen verfolgt die MPBetreibV auch die Ziele dieses Rechtssystems: die Sicherheit, Leistungsfähigkeit und Wirksamkeit und damit auch die Sicherheit der Patienten sicherzustellen.
d) Was fordert die MPBetreibV?
Das zweite Kapitel dieses Artikels beschreibt die Anforderungen.
e) Wer ist für die Überwachung der MPBetreibV verantwortlich?
Die Überprüfung der Einhaltung der MPBetreibV obliegt den Landesbehörden der einzelnen Bundesländer.
2. MPBetreibV: Die wichtigsten Anforderungen
a) Anforderungen an das Betreiben
Die Medizinprodukte-Betreiberverordnung stellt Anforderungen an das Betreiben. Darunter zählt sie u. a.:
- Instandhaltung von Produkten (§ 7 MPBetreibV)
- Aufbereitung von Produkten (§ 8 und § 9 MPBetreibV)
- Einweisung der Benutzer (§ 4, § 11 und § 17 MPBetreibV)
- Sicherheitstechnischen Kontrollen STK (§ 12 MPBetreibV) (s. u.)
- Führen eines Medizinproduktebuchs (§ 13 MPBetreibV)
- Führen eines Bestandsverzeichnisses (§ 14 MPBetreibV) (s. u.)
- Messtechnische Kontrollen MTK (§ 15 MPBetreibV) (s. u.)
- IT-Sicherheitsüberprüfungen (§ 17 MPBetreibV)
Ein weiterer Artikel zur Instandhaltung gibt konkrete Hinweise, was bei der Wartung und Instandhaltung von Medizinprodukten zu beachten ist.
b) Anforderungen an den Betreiber und Benutzer
Es gibt Anforderungen an den Betreiber bzw. die Personen selbst, z. B. in § 4 MPBetreibV:
Produkte dürfen nur von Personen betrieben oder benutzt werden, die die dafür erforderliche Ausbildung oder Kenntnis und Erfahrung besitzen.
§ 4 (2) MPBetreibV
Diese Forderungen richten sich nicht nur an Ärzte. Ärzte verfügen auch nicht notwendigerweise über alle Kompetenzen, um Medizinprodukte zu betreiben. Beispielsweise sind bei Software auch Tätigkeiten notwendig wie das Update des Betriebssystems, der Laufzeitumgebung, das Aktualisieren von Firewalls und Antivirus-Programmen, um den sicheren Betrieb sicherzustellen.
c) Anforderungen an Software und vernetzte Produkte
Die Sicherheit von Patienten, Benutzern und Dritten steht auch bei der MPBetreibV im Mittelpunkt. Explizit für Produktkombinationen, wozu auch mit einem Netzwerk verbundene Produkte zählen, schreibt sie:
Miteinander verbundene Produkte sowie Produkte, die mit anderen Gegenständen verbunden sind, dürfen nur betrieben und benutzt werden, wenn sie dafür in dieser Kombination unter Berücksichtigung der Zweckbestimmung und der Sicherheit der Patienten, Benutzer, Beschäftigten oder Dritten geeignet sind.
§ 4 (4) MPBetreibV
Des Weiteren adressiert die MPBetreibV an mehreren Stellen Software und vernetzte Produkte explizit:
- Nach jeder Softwareaktualisierung, die die Handhabung der Software nicht nur geringfügig ändert, ist eine Einweisung in die ordnungsgemäße Handhabung des Produkts erforderlich (§ 4 MPBetreibV).
- Für vernetzte Produkte müssen bei der Verbindung mit einem Netzwerk die Anforderungen des Herstellers an die IT-Sicherheit beachtet werden (§ 4 MPBetreibV).
- Bei Software umfasst die Instandhaltung auch die Installation sicherheitsrelevanter Softwareupdates (§ 7 MPBetreibV).
- Für Software der Klassen IIb und III nach MDR sowie der Klassen C und D nach IVDR gelten ab August 2025 besondere Vorschriften, wie die erforderliche Prüfung der Installation sowie das Durchführen von IT-Sicherheitsüberprüfungen (§ 17 MPBetreibV).
Anforderungen an das Risikomanagement
Betreiber dieser „verbundenen Produkte“ sollten weitere spezielle Regelungen beachten, um die Konformität mit den Anforderungen der MPBetreibV zu gewährleisten. Beispielsweise wendet sich die Norm DIN EN IEC 80001-1 ebenfalls an die Betreiber. Sie stellt Anforderungen, wie jene an das Risikomanagement für IT-Netzwerke, die Medizinprodukte beinhalten, anwenden sollen.
Die DIN EN IEC 80001-1 ist zwar gesetzlich nicht gefordert, jedoch schreibt der branchenspezifische Sicherheitsstandard (B3S) der Deutschen Krankenhausgesellschaft (DKG), dass die Anforderungen der DIN EN 80001-1 für den Einsatz von Medizingeräten in medizinischen IT-Netzwerken berücksichtigt werden sollen.
Exkurs
Seit 2022 sind alle Krankenhäuser in Deutschland durch § 75c, mittlerweile § 391 des SGB V verpflichtet, nach dem Stand der Technik angemessene organisatorische und technische Vorkehrungen zur Sicherstellung der IT-Sicherheit ihrer informationstechnischen Systeme, Komponenten oder Prozesse zu treffen. Die Krankenhäuser können diese Verpflichtung erfüllen, wenn sie einen branchenspezifischen Sicherheitsstandard für die informationstechnische Sicherheit der Gesundheitsversorgung im Krankenhaus in der jeweils gültigen Fassung anwenden.
Lesen Sie mehr zum Thema Risikomanagement im Krankenhaus und den Forderungen der MPBetreibV sowie zur DIN EN IEC 80001-1.
d) Eintragungen in das Bestandsverzeichnis
Gesetz und Definition
Die MPBetreibV schreibt dazu:
Der Betreiber hat für alle aktiven nicht implantierbaren Produkte der jeweiligen Betriebsstätte ein Bestandsverzeichnis zu führen. Die Aufnahme in ein Verzeichnis, das auf Grund anderer Vorschriften geführt wird, ist zulässig.
§ 14 (1) MPBetreibV
Was aktive Medizinprodukte sind, definiert die MDR:
Ein Produkt, dessen Betrieb von einer Energiequelle mit Ausnahme der für diesen Zweck durch den menschlichen Körper oder durch die Schwerkraft erzeugten Energie abhängig ist und das mittels Änderung der Dichte oder Umwandlung dieser Energie wirkt. Ein Produkt, das zur Übertragung von Energie, Stoffen oder anderen Elementen zwischen einem aktiven Produkt und dem Patienten eingesetzt wird, ohne dass dabei eine wesentliche Veränderung von Energie, Stoffen oder Parametern eintritt, gilt nicht als aktives Produkt.
Software gilt ebenfalls als aktives Produkt;
Beispiele für Produkte, die im Bestandsverzeichnis geführt werden müssen
Medizinprodukte, die in das Bestandsverzeichnis aufgenommen werden müssen, sind:
- Medizinisch-elektrische Geräte wie Röntgengeräte, CTs, Ultraschallgeräte, Patientenmonitore, elektrische Blutdruckmessgeräte, Defibrillatoren
- Software als Medizinprodukt, beispielsweise PACS, die meisten Radiologie-Informations-Managementsysteme (RIS) und Patientendaten-Managementsysteme (PDMS), sofern es sich um Medizinprodukte handelt
- Elektrisch betriebene Rollstühle
- Aktive Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation von Medizinprodukten vorgesehen sind
- Laborgeräte, sofern es sich um In-vitro-Diagnostika handelt
Streng genommen müssten sogar elektrische Fieberthermometer aufgenommen werden.
Auch Zubehör muss in das Bestandsverzeichnis aufgenommen werden.
Beispiele für Produkte, die NICHT im Bestandsverzeichnis geführt werden müssen
Hingegen unterliegen diese Produkte nicht der Pflicht, in ein Bestandsverzeichnis aufgenommen zu werden:
- Chirurgische Instrumente
- Blutdruckmessgeräte und Stethoskope, die ohne Strom genutzt werden
- Krankenhaus-Informationssysteme (wenn diese kein Medizinprodukt sind)
- Rollstühle, die ohne Strom betrieben werden
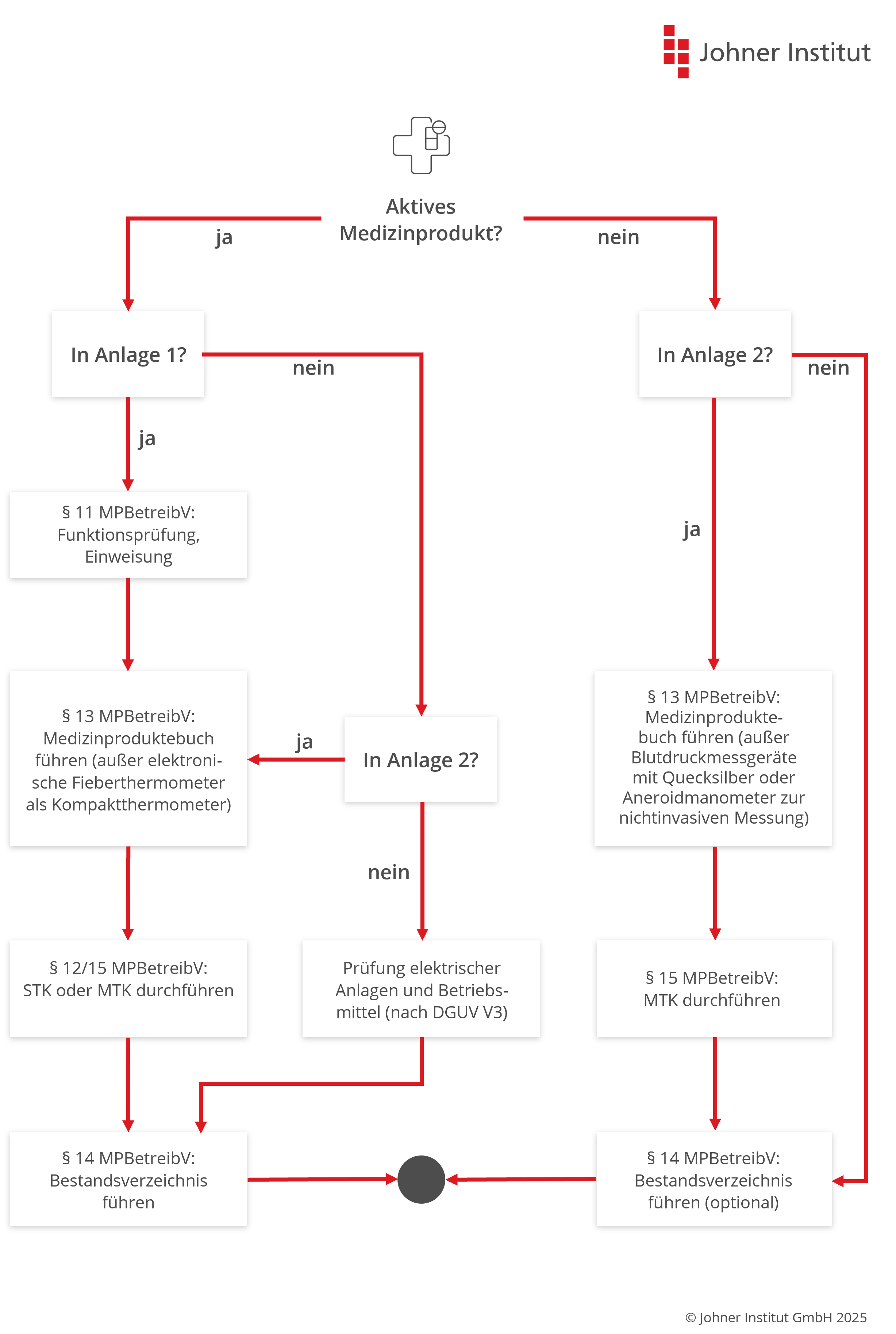
e) Sicherheitstechnische Kontrollen (STK)
Anforderungen an sicherheitstechnische Kontrollen
Sicherheitstechnische Kontrollen (STK) haben das Ziel, die Sicherheit von Produkten zu überprüfen und sicherzustellen, um Gefährdungen für Patienten, Benutzer und Dritte zu minimieren.
Die STK sind aber nur für Produkte nach Anlage 1 vorgeschrieben. Das sind im Wesentlichen kritische aktive, nicht implantierbare Produkte wie Kernspingeräte, Stimulatoren, Beatmungsgeräte oder Geräte zur Lithotripsie.
Manche Hersteller verlangen in der Gebrauchsanweisung ihrer Medizinprodukte die Durchführung sicherheitstechnischer Kontrollen – auch für Produkte, die nicht in Anlage 1 aufgeführt sind. Unter dem Begriff „sicherheitstechnische Kontrolle“ verstehen sie dabei Prüfungen im Rahmen der Instandhaltung (§ 7 MPBetreibV). Diese sind zu beachten, jedoch strikt von den vorgeschriebenen STK nach § 12 zu unterscheiden.
Anlage-I-Medizinprodukte
Die Betreiberverordnung listet in der Anlage insgesamt 10 Produkttypen:
- Nicht implantierbare aktive Produkte zur Erzeugung und Anwendung elektrischer Energie zur unmittelbaren Beeinflussung der Funktion von Nerven und/oder Muskeln beziehungsweise der Herztätigkeit einschließlich Defibrillatoren
- Nicht implantierbare aktive Produkte zur intrakardialen Messung elektrischer Größen oder Messung anderer Größen unter Verwendung elektrisch betriebener Messsonden in Blutgefäßen beziehungsweise an freigelegten Blutgefäßen
- Nicht implantierbare aktive Produkte zur Erzeugung und Anwendung jeglicher Energie zur unmittelbaren Koagulation, Gewebezerstörung oder Zertrümmerung von Ablagerungen in Organen
- Nicht implantierbare aktive Produkte zur unmittelbaren Einbringung von Substanzen und Flüssigkeiten in den Blutkreislauf unter potenziellem Druckaufbau, wobei die Substanzen und Flüssigkeiten auch aufbereitete oder speziell behandelte körpereigene sein können, deren Einbringen mit einer Entnahmefunktion direkt gekoppelt ist
- Nicht implantierbare aktive Produkte zur maschinellen Beatmung mit oder ohne Anästhesie, mit Ausnahme von Schlafapnoe-Therapiegeräten
- Nicht implantierbare aktive Produkte zur Diagnose mit bildgebenden Verfahren nach dem Prinzip der Kernspinresonanz
- Nicht implantierbare aktive Produkte zur Therapie mit Druckkammern
- Nicht implantierbare aktive Produkte zur Therapie mittels Hypothermie
- Säuglingsinkubatoren
- Externe aktive Komponenten aktiver Implantate
Inspektionen und Wartungen
Die MPBetreibV spricht in § 7 von Inspektionen und Wartungen:
„Wartungen und Inspektionen, die für die Gewährleistung des sicheren und ordnungsgemäßen Produktbetriebs erforderlich sind, sowie Instandsetzungen zur Wiederherstellung der Funktionsfähigkeit eines Produktes. Die Angaben des Herstellers sind dabei zu berücksichtigen.“
§ 7 (2) MPBetreibV
Umgekehrt können Hersteller die Betreiber nicht von den Pflichten zu STK befreien. Auch von der Gefährdungsbeurteilung und sicherheitstechnischen Bewertung (TRBS 1111) können die Hersteller die Betreiber nicht entbinden. Vielmehr sollten die Betreiber die Angaben der Hersteller für diese Beurteilung nutzen.
Sicherheitstechnische Kontrollen sind spätestens alle zwei Jahre mit Ablauf des Monats durchzuführen, in dem die Inbetriebnahme des Produkts erfolgte oder die letzte sicherheitstechnische Kontrolle durchgeführt wurde. STK sind von qualifizierten Personen durchzuführen und zu dokumentieren.
f) Messtechnische Kontrollen (MTK)
Messtechnische Kontrollen (MTK) haben das Ziel, die Messgenauigkeit von Produkten sicherzustellen. Die MPBetreibV nennt (in Anlage 2) die Produkte, die einer MTK unterzogen werden müssen. Dazu zählen unter anderem medizinische Elektrothermometer, nichtinvasive Blutdruckmessgeräte, Tretkurbelergometer und verschiedene Dosimeter.
Im Gegensatz zur STK, die spätestens alle zwei Jahre durchgeführt werden muss, gibt es bei der MTK je nach Art des Produkts unterschiedliche Nachprüffristen, die in Anlage 2 festgelegt sind. Die Fristen beginnen mit Ablauf des Jahres, in dem das Produkt in Betrieb genommen oder die letzte MTK durchgeführt wurde.
Auch die MTK sind von qualifizierten Personen durchzuführen und zu dokumentieren.
Hersteller dürfen den Betreibern kürzere, aber keine längeren Prüffristen vorschreiben. Sie können die Betreiber nicht von der Pflicht zu MTK befreien.
g) Einweisung in Medizinprodukte
Die MPBetreibV fordert eine Einweisung in die Handhabung der Medizinprodukte in § 4. Die Verordnung erlaubt Ausnahmen nur, falls
- für das Produkt keine Einweisung erforderlich ist, oder
- eine Einweisung in ein baugleiches Produkt erfolgt ist.
Ob eine Einweisung erforderlich ist, bestimmen die MDR bzw. IVDR in den Anhängen I:
Eine Gebrauchsanweisung ist für Produkte der Klassen I und IIa ausnahmsweise entbehrlich, wenn eine sichere Anwendung dieser Produkte ohne Gebrauchsanweisung gewährleistet ist und sofern an anderer Stelle dieses Abschnitts nichts anderes angegeben ist.
MDR, Anhang I, Abschnitt 23.1 d)
Bei aktiven nicht implantierbaren Produkten verlangt die MPBetreibV eine Dokumentation.
Auch für Software gibt es eine Besonderheit: Nach Software-Updates, die „die Handhabung nicht nur geringfügig ändern“ sind die Betreiber zu einer erneuten Einweisung verpflichtet.
3. Betreiber im Sinne der Medizinprodukte-Betreiberverordnung (MPBetreibV)
Definition
Die aktuelle MPBetreibV (Februar 2025) definiert den Begriff „Betreiber“ wie folgt:
Betreiber eines Produktes ist jede natürliche oder juristische Person, die für den Betrieb der Gesundheitseinrichtung verantwortlich ist, in der das Produkt durch dessen Beschäftigte betrieben oder benutzt wird. Abweichend von Satz 1 ist Betreiber eines Produktes, das im Besitz eines Angehörigen der Heilberufe oder des Heilgewerbes ist und von diesem zur Verwendung in eine Gesundheitseinrichtung mitgebracht wird, der betreffende Angehörige des Heilberufs oder des Heilgewerbes. Als Betreiber gilt auch, wer außerhalb von Gesundheitseinrichtungen Produkte zur Benutzung bereithält.
Bewertung
Unter Betreiber versteht man intuitiv eher Institutionen als Personen. Typische Betreiber sind Krankenhäuser, Arztpraxen, diagnostische Labore und medizinische Versorgungszentren. Abweichend davon gelten auch Angehörige der Heilberufe oder des Heilgewerbes, wie Belegärzte, als Betreiber, wenn sie ein Medizinprodukt zur Verwendung in einer Gesundheitseinrichtung mitbringen. Ebenso gilt als Betreiber, wer Medizinprodukte außerhalb von Gesundheitseinrichtungen zur Benutzung bereithält, etwa einen Defibrillator in öffentlichen Einrichtungen.
Betreiber im SaaS-Kontext
Hingegen handelt es sich bei einem SaaS-Provider (SaaS = Software as a Service) einer Medical Device Software (MDSW) nicht um einen Betreiber im Sinne der MPBetreibV, da die MDSW weder auf dessen unternehmensseitigem Server zur Benutzung bereitgehalten noch in einem sonstigen Betrieb bereitgestellt wird. Die MDSW wird stattdessen in einem begrenzten Kreis von (Gesundheits-)Einrichtungen auf Basis vertraglicher Vereinbarungen zur Benutzung zugänglich gemacht, wobei die Entscheidung über den Einsatz der MDSW vollständig beim Kunden verbleibt.
Der SaaS-Provider ist nicht für den medizinischen Kontext oder dessen Umstände verantwortlich und daher kein Betreiber im Sinne der MPBetreibV. Er ist vergleichbar mit einem Dienstleister, der den technischen Betrieb medizinischer Geräte übernimmt, ohne selbst Betreiber zu sein.
Weitere Definitionen
Weil die MPBetreibV auch für Benutzer und Versorgende relevant ist, hier noch die Definition dieser Begriffe:
Benutzer im Sinne dieser Verordnung ist, wer ein Produkt im Anwendungsbereich dieser Verordnung am Patienten einsetzt.
Versorgender ist, wer aufgrund einer gesetzlichen oder vertraglichen Verpflichtung gegenüber dem Patienten Produkte bereitzustellen hat.
4. MDR/IVDR und MPBetreibV
Die MDR und die IVDR geben in Europa den rechtlichen Rahmen für Medizinproduktehersteller vor. Sie stellen auch einige Anforderungen an die Gesundheitseinrichtungen, z. B. bezüglich:
- Eigenherstellung
- Wiederaufbereitung
- UDI
MDR und IVDR regeln aber nur in sehr eingeschränktem Umfang die Benutzung und den Betrieb der Medizinprodukte. Dies ist die Aufgabe der Mitgliedsstaaten.
Nationale Verordnungen bedürfen immer eines Gesetzes, das eine entsprechende „Verordnungsermächtigung“ enthält. Im Falle der MPBetreibV ist das der § 88 im MPDG.
Lesen Sie mehr zu den Anforderungen der MDR und IVDR an die Betreiber (Gesundheitseinrichtungen) und zum MPDG.
5. Änderungen an der MPBetreibV
a) Änderungen zum 01.01.2017
Die Medizinprodukte-Betreiberverordnung (MPBetreibV) wurde im September 2016 überarbeitet. Die neue Version trat am 01.01.2017 in Kraft. Zu den Änderungen der Verordnung zählen:
- Die Verordnung enthält nun die Definition des Begriffs Betreiber.
- Anwender müssen sich nicht nur vom Funktionieren von Medizinprodukten überzeugen, sondern auch von dem Funktionieren verbundener Produkte. Der zunehmenden Vernetzung soll hier Rechnung getragen werden. Es bleibt unklar, wie Anwender diese Forderung erfüllen sollen.
- Gesundheitseinrichtungen mit mehr als 20 Mitarbeitenden müssen einen zentralen Ansprechpartner benennen und bereitstellen, den Beauftragten für Medizinproduktesicherheit. Die Anforderungen sind niedriger als jene an die Sicherheitsbeauftragten bei Herstellern.
- Eine Einweisung wird für alle Medizinprodukte gefordert. Ein bloßes „learning on the job“ ist nicht gestattet.
- Die sicherheitstechnischen Kontrollen sind nur noch für Produkte gefordert, die in Anlage 1 gelistet sind, es sei denn, der Hersteller fordert diese (mehr dazu weiter oben).
- Die messtechnischen Kontrollen sind nur noch für Produkte gefordert, die in Anlage 2 gelistet sind.
b) Änderungen im Jahr 2020/2021
Die Medizinprodukte-EU-Anpassungsverordnung (MPEUAnpV) hat die Medizinprodukte-Betreiberverordnung (MPBetreibV) geändert.
Laden Sie sich hier die MPEUAnpV herunter. Die Änderungen der MPBetreibV finden Sie auf Seite 18 f.
Die MPBetreibV wurde u. a. in den folgenden Punkten geändert:
- Anwendungsbereich: jetzt (auch) MDR und IVDR
- Verantwortlichkeiten der Behörde
- Regelungen für Einmalprodukte und deren Aufbereitung
- Referenzen
- Pflichten im Kontext der Implantationsausweise
Viele Änderungen sind formaler Natur. So mussten die Verweise auf die EU-Richtlinien in Verweise auf die EU-Verordnungen geändert werden.
Sie finden hier eine Gegenüberstellung der alten und neuen MPBetreibV.
c) Änderungen im Februar 2025
Am 20. Februar 2025 ist eine neue Version der MPBetreibV in Kraft getreten, die diverse Änderungen im Vergleich zur vorherigen Version mit sich bringt.
Anlass der Änderungen
Hauptanlass vieler Änderungen ist die zunehmende Digitalisierung im Gesundheitswesen und der damit einhergehende vermehrte Einsatz von Medizinprodukte-Software, der Sorgen zu bereiten scheint, wie die Erwägungsgründe im Referentenentwurf des Bundesministeriums für Gesundheit (BMG) aus November 2023 erkennen lassen:
Deutlich wurde, dass die zunehmende Digitalisierung neuer, auf ihre Besonderheiten zugeschnittener Regelungen bedarf. Es ist zu beobachten, dass Gesundheitseinrichtungen auch vermehrt auf den Einsatz von Medizinprodukte- Software bei den Behandlungen von Patienten zurückgreifen. Dies birgt verschiedene Risiken für das sichere Funktionieren der Produkte […]
Referentenentwurf auf Seite 1
Änderungen
Neben den Änderungen zur Software gibt es zahlreiche weitere Anpassungen. Im Folgenden sind einige der geänderten Anforderungen aufgeführt:
- Der Anwendungsbereich der Verordnung wurde auf alle in § 3 Nr. des MPDG genannten Produkte ausgeweitet und umfasst nun auch Produkte nach Anhang XVI der MDR ohne medizinischen Verwendungszweck (gültig ab August 2025).
- Die Begriffe „Anwender“ bzw. anwenden“ wurden durch „Benutzer“ bzw. „benutzen“ in der Verordnung ersetzt.
- Der Begriff des „Versorgenden“ wurde eingeführt.
- Für Software ist nach jeder Installation von Softwareaktualisierungen, die die Handhabung der Software mehr als geringfügig ändern, eine Einweisung erforderlich.
- Die Aufbereitung von Produkten zur Mehrfachanwendung und zur Einzelanwendung wurde getrennt geregelt (neuer § 9).
- Eine Verpflichtung zur Kennzeichnung der Produkte nach einer durchgeführten STK wurde eingefügt.
- Die im Bestandsverzeichnis zu führenden Angaben wurden angepasst.
- Besondere Pflichten bei bestimmter Software (neuer § 17), z. B. das Durchführen von IT-Sicherheitsüberprüfungen, wurden eingeführt (gültig ab August 2025).
Gerne stellen wir Ihnen eine kostenlose Übersicht über alle inhaltlichen Änderungen der neuen MPBetreibV zur Verfügung. Melden Sie sich bei Interesse einfach über unser Kontaktformular!
Änderungshistorie
- 2025-03-28: Abschnitt 2.e) erweitert, Abschnitt 2.g) ergänzt
- 2025-03-04: Artikel komplett überarbeitet aufgrund neuer MPBetreibV 2025
- 2025-02-13: Artikel weitestgehend überarbeitet und im Hinblick auf neue geplante Änderungen aktualisiert
- 2024-05-25: Boxen mit Warnungen und weiterführenden Informationen in den Kapiteln 1 a) und 2 a) ergänzt
- 2023-11-29: In Kapitel 7 c) die geplanten Änderungen an der MPBetreibV aufgenommen
- 2023-05-04: Artikel aktualisiert, FAQ ergänzt, Verweise auf die alte MPBetreibV korrigiert
- 2021-05-31: Artikel weitgehend überarbeitet, Historie gelöscht, Kapitel 3 eingefügt, Kapitel 6 ergänzt



Die Zuständigkeit der MPBetreibV hat doch nichts direkt mit dem MPG zu tun welches als MPG jedoch aufs Produkt bezogen ist.
Die MPBetreibV dient doch ausschließlich dem Schutz von Betrieben und somit deren Arbeitnehmern.
Hiervon ist bei einer normalen Homecare Versorgung, aufgrund einer ärztlichen VO und Kostenübernahem durch die GKV gem. SGB V, wie z. B. Inahalationsgerät, Blutdruckmeßgerät, etc., das MPBetreibV gar nicht davon tangiert. Der Patient nutzt das Medizinprodukt in eigener Sachherrschaft und Verantwortung ohne Arbeitgebereigenschaft, so dass nur das MPG greift da das Med.-Gerät zum therapeutischen Einsatz im privaten Bereich kommt und somit das MPG mit der Einweisungspflicht greift. Ansonsten müssten ja alle Verkäufe über Laden, Internet etc. gestoppt werden, weil es hierbei zu keiner Anwendungsberatung und Einweisung gem. MPG kommt.
Die MPBetreibV ist eine durch §37 autorisierte Ergänzung des MPG. Beide haben somit etwas miteinander zu tun.
Die MPBetreibV gilt (wie das MPG) ganz explizit auch dem Schutz von Patienten – nicht nur dem der Arbeitnehmer. Die Arbeitbeitsschutzgesetze gelten zusätzlich – wie die MPBetreibV klarstellt.
Eine Homecare-Versorgung kann durchaus der MPBetreibV unterliegen. Den Begriff des Betreibens klärt die MPBetreibV in §2.
Eine Apotheke ist kein Betreiber.
Ob eine Einweisungspflicht besteht, hängt von der vertraglichen Konstellation ab.
Ein schwieriges Thema aber hier einmal quergedacht…kein Mainstream
Im Gesetz über Medizinprodukte (Medizinproduktegesetz – MPG) regelt der §14 MPG den grundsätzlichen Schutzanspruch, ohne den Begriff des Betreibers, für alle Beteiligten im Mangel- u. Gefahrenausschluss sowie unter Einbeziehung des § 37 Abs. 5:
MPG § 14 Tätigkeiten im Zusammenhang mit Medizinprodukten
Medizinprodukte dürfen nur nach Maßgabe der Rechtsverordnung nach § 37 Absatz 5 betrieben und angewendet werden. Medizinprodukte dürfen nicht betrieben und angewendet werden, wenn sie Mängel aufweisen, durch die Patienten, Beschäftigte oder Dritte gefährdet werden können.
Die MPBetreibV ist eine Ableitung aus dem MPG § 37 Verordnungsermächtigungen, anbei der Auszug aus dem übergeordneten(Höherem) Gesetz:
MPG § 37 (5) Das Bundesministerium für Gesundheit wird ermächtigt, durch Rechtsverordnung
1. Anforderungen an das Errichten, Betreiben, Anwenden und Instandhalten von Medizinprodukten festzulegen, Regelungen zu treffen über die Einweisung der Betreiber und Anwender, die sicherheitstechnischen Kontrollen, Funktionsprüfungen, Meldepflichten und Einzelheiten der Meldepflichten von Vorkommnissen und Risiken, das Bestandsverzeichnis und das Medizinproduktebuch sowie weitere Anforderungen festzulegen, soweit dies für das sichere Betreiben und die sichere Anwendung oder die ordnungsgemäße Instandhaltung notwendig ist“
aus diesem Absatz ergibt sich eben nicht explizit die Zuständigkeit sprich Anwendungsbereich der MPBetreibV für Patienten, denn diese Zuständigkeit/Geltungsbereich ist eben nicht im im MPG § 14 weiter geregelt, sondern wie folgt in der MPBetreibV:
weiterer Auszug nunmehr aus der MPBetreibV: §2 Begriffsbestimmungen
(2) Betreiber eines Medizinproduktes ist jede natürliche oder juristische Person, die für den Betrieb der Gesundheitseinrichtung verantwortlich ist, in der das Medizinprodukt durch dessen Beschäftigte betrieben oder angewendet wird. Abweichend von Satz 1 ist Betreiber eines Medizinproduktes, das im Besitz eines Angehörigen der Heilberufe oder des Heilgewerbes ist und von diesem zur Verwendung in eine Gesundheitseinrichtung mitgebracht wird, der betreffende Angehörige des Heilberufs oder des Heilgewerbes. Als Betreiber gilt auch, wer außerhalb von Gesundheitseinrichtungen in seinem Betrieb oder seiner Einrichtung oder im öffentlichen Raum Medizinprodukte zur Anwendung bereithält.
Genau hier wird explizit der Patient ausgeschlossen und nicht aufgeführt, sondern nur auf Gewerbsmäßige Einrichtungen abgehoben. Selbst im nächsten Absatz wird nur Bezug auf den Anwendungsbereich als Anwender ausschließlich auf diese Verordnung, MPBetreibV, für diesen Geltungsbereich Bezug genommen. Siehe Auszug:
(3) Anwender ist, wer ein Medizinprodukt im Anwendungsbereich dieser Verordnung am Patienten einsetzt.
Natürlich kann nicht nur, sondern ist eine Apotheke, Praxis, etc. oder auch andere Gewerbe ein Betreiber sein sobald diese eben ein Medizinprodukt wie z. B. ein Blutdruckmessgerät in ihrem Bereich einsetzen, egal ob kostenlos oder gegen Bezahlung.
Eine Einweisungspflicht ist bei einem zur Therapie verordneten Medizinprodukt meiner Auffassung nach immer gegeben und kann nicht Gegenstand vertraglicher Konstruktionen sein da ansonsten das MPG ausgehebelt würde und zur Farce verkämme. Des Weiteren ergeben sich die weiteren Regelungen und Sorgfaltspflichten aus dem SGB 5, BGB und den Krankenkassenverträgen.
Hallo Herr Johner,
danke für den guten Artikel zum Thema.
Eine Frage bleibt:
Inwiefern lesen Sie aus dem § 7 I MPBetreibV eine STK-Pflichtigkeit für MP außerhalb der Anlage 1? Inspektion und Wartung erfüllen für mich begriffsauslegend lediglich sicht-, funktions- und sicherheitstechnische Aspekte mit Abgrenzung zu etwaigen Messpflichten.
Ohne abschließende Regelung der Anlagen wäre dann ja die Anlage 1 völlig redundant.
BG,
Gerwin S.
Sehr geehrter Herr S. danke für Ihre Nachricht.
Ich bin nicht sicher, ob ich Ihre Frage verstehe. Können Sie mir helfen?: Mit welchem Satz im Artikel haben Sie Schwierigkeiten? Wie sollte dieser korrekterweise lauten.
Sehr gerne bessere ich nach, bedarf nur Ihres Hinweises, wo ich ansetzen soll.
Viele Grüße, Christian Johner
Hallo,
zu Anfang schreiben Sie „es sei denn Hersteller fordert diese (mehr dazu weiter unten)“.
Nach meinem Verständnis der Novelle und der Anlagen 1 und 2 ist die Instandhaltung i.S.d. § 7 MPBetreibV schon begrifflich ein weniger als eine STK und MTK. Gerade damit nicht jeder Hersteller eines nicht kritischen Medizinproduktes eine STK per Gusto anordnen kann, sehe ich ich die Instandhaltung, auch nach Herstellerangaben, als eine qualifizierte Funktionellen Trolle. Nicht mehr. Oder gibt es zum Umfang des § 7 schon seriöse Kommentarliteratur?
MfG
Sehr geehrter Herr Schroer,
danke für Ihre Anmerkung und Ihre Frage.
Einem Hersteller ist es immer offen, mehr zu verlangen, als die das Gesetz tut. Hersteller vermeiden das nur, um die Akzeptanz der Produkte zu erhöhen. Aber ein Verbot über das Gesetz hinausgehender Vorschriften gibt es nicht.
Eine „seriöse Kommentarliteratur“ zum §7 ist mir nicht bekannt.
Herzliche Grüße, Christian Johner
Hallo Herr Johner,
natürlich ist vieles möglich – ich denke aber, dass die abschließende Regelung der Anlagen 1 und 2 (wenigstens) starke Indizwirkung entfaltet für eine „STK-Marktbereinigung“ der Hersteller von Medizin- und Rehatechnik.
Im prüf- und plakettenscharfen Deutschland muss auch deutlich werden (können), dass nicht kritische Medizinprodukte nicht jenseits des durchschnittlichen Arbeits- und Funktionsschutzes zu prüfen sind.
Und natürlich wird der Fachhandel auch durch sein Bestell- und Kooperationsverhalten die Medizinproduktbranche mitgestalten (müssen).
Danke für den freundlichen Diskurs!
MfG,
G. Schroer
Sehr geehrter Herr Prof. Dr. Johner,
mir ergründet sich aus den Kommentaren und Fragen leider immer noch nicht folgender Sachverhalt.
Wir sind vom Fachhandel ( Sanitätshaus), das vorausgeschickt.
Die Krankenkassen haben oftmals in den geschlossenen Verträgen nur einen kurzen Hinweis… die Betreiberpflichten werden auf den Vertragspartner ( Sanitätshaus) übertragen.
Dies betrifft alles im Zusammenhang der Hilfsmittelversorgung . Beratung Anpassung Aufbereitung Lieferung Einweisung spätere Reparatur Instandhaltung STK und Wartung.
Nach unserer Ansicht betrifft das alle aktiven Medizinprodukte.
Auch über die Anlage 1 und 2 hinaus . Ist das richtig?
Wenn ja. Betrifft das auch solche Produkte die vom Hersteller als wartungsfrei in der Bedienungsanleitung beschrieben werden? z. B. CPAP Geräte.
Hierzu kommen oft Fragen vom Kostenträger( Krankenkasse) …. der uns ja vertraglich verpflichtet hat…. mit dem Hinweis zahle ich nicht …, das Produkt ist wartungsfrei .
Über eine Antwort würde ich mich sehr freuen.
Herzliche Grüße
Rita Becher
Sehr geehrte Frau Bescher,
besten Dank für Ihr Nachhaken. Ich bin nicht ganz sicher, ob ich Ihre Frage wirklich verstanden haben. Daher antworte ich mal auf „gut Glück“:
§11 MPBetreibV sagt, dass die Betreiber „für die in der Anlage 1 aufgeführten Medizinprodukte sicherheitstechnische Kontrollen nach den allgemein anerkannten Regeln der Technik und nach Satz 2 oder Satz 3 durchzuführen oder durchführen zu lassen“ haben.
Diese Anlage 1 führt zwar nur aktive Medizinprodukte auf. Aber d.h. nicht, dass alle aktiven Medizinprodukte einer STK unterworfen werden müssen.
Die Aussage eines Herstellers, dass ein Produkt wartungsfrei sein, ändert daran nichts. Schließlich kann ein wartungsfreies Produkt auch defekt werden. Genau das soll bei einer STK entdeckt werden.
Viele Grüße
Christian Johner
Hallo Herr Johner,
ich arbeite an einer öffendlichen Schule wo eine staatlich anerkannte Ausbildung zum medizintechnische Assistenten und Medizintechniker angeboten wird. Wir besitzen sehr viele Medizingeräte, wo die Schüler bzw. Studierende die Funktionsprüfungen bzw. Instandhaltungen und sogar die richtige Anwendung dieser Geräte erlernen. Meine Frage an Sie, im §2 MP BetreibV Absatz (4) wird erklärt was eine Gesundheitseinrichtung im Sinne der Verordung ist. …sonstige dazu befugte Personen berufsmäßig betrieben oder angewendet werden.
Sind wir als Schule im Sinne der Verordnung eine Gesundheitseinrichtung? Im Rahmen der Betiebssicherheitsverordnung stehen gerade Prüfungen an und wir sind uns nicht sicher, da es ja um Schulungsgeräte handelt, ob wir nach MPBeteibV handeln bzw. prüfen müssen.
Mit freundlichen Grüßen
P. Nieleck
Sie sind keine Gesundheitseinrichtung. Die Definition dieses Begriffs in der MPBetreibV ist eigentlich klar, aber sie ist nicht so eindeutig, wie Sie in der MDR nun ist:
36. „Gesundheitseinrichtung“ bezeichnet eine Organisation, deren Hauptzweck in der Versorgung oder Behandlung von Patienten oder der Förderung der öffentlichen Gesundheit besteht;
Also Entspannung!
Sehr geehrter Herr Johner.
Im Rahmen einer Erstellung eines Gutachtens, als Sachverständiger für Medizinprodukte, bin ich mit einer ungewöhnlichen Konstellation konfrontiert worden:
Ein Hersteller eines etablierten Produktes bewirbt und vertreibt seine Produkte als offensichtliche Medizinprodukte. Auf Anfrage nach der EU Konformitätserklärung erhielten wir die Aussage, dass es sich bei dem Produkt nicht um ein Medizinprodukt handeln würde und daher auch keine EU Konformitätserklärung erhältlich sei.
Etwas überrascht von dieser Haltung, haben wir diese Erkenntnisse an das BfArM und die zuständigen Gewerbeaufsichtsämter, der uns bekannten Betreiber der Produkte, weitergeleitet, insbesondere weil uns Erkenntnisse über eine akute Patientengefährdung vorliegen. Der gesamte Vorgang begann vor ca. 2 Jahren! und hat bisher nicht dazu geführt, dass die Bewerbung und der Vertrieb der Produkte geändert wurde.
Da dies u.a. nicht kompatibel mit den Vorgaben der MPBetreibV ist, möchte ich Sie um die Information bitten, wer eigentlich für die Überwachung der Umsetzung der MPBetreibV zuständig ist.
Dieser Fall führt aus meiner Sicht unsere gesamten Bemühungen, nach einer korrekten Umsetzung der Anforderungen, ad absurdum, wenn es problemlos möglich ist, unter den Augen der Behörden ein offensichtliches Medizinprodukt bzw. eine gesamte Produktpalette ohne jegliche Nachweise der Konformität auf dem Markt zu etablieren.
Für Ihre Unterstützung im voraus besten Dank.
Grüsse
Thomas Castner
Sehr geehrter Herr Dr. Castner,
danke für die spannende Frage: Wir haben hier zwei Themen: Das eine ist die offensichtliche unrechtmäßige Inverkehrbringung. Hierfür sind die Landesbehörden und ihre nachgelagerten Behörden wie Gewerbeaufsichtsämter oder Regierungspräsidien zuständig.
Diese Behörden sind meist auch für die Aufsicht der Krankenhäuser zuständig. Allerdings sind das oft andere Abteilungen.
Das Problem mit der Inaktivität dieser Behörden überrascht mich nicht. Ich warte auch auf eine Antwort seit letztem November. Hier hilft nur eine Dienstaufsichtsbehörde.
Traurig aber wahr.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
könnten Sie kurz auf die Frage STK für AED eingehen.
Hier besonders auf die Definition des „öffentlichen Raums“?
Grüße und Dank,
Jürgen C.
Sehr geehrter Herr C.
Sie finden die Antwort auf diese Frage hier: https://defi.de/medizinprodukte-betreiberverordnung/#was-ist-oeffentlicher-raum
Ehre wem Ehre gebührt, daher wollte ich Ihnen die wie ich finde beste Quelle nennen.
Mit herzlichen Grüßen, Christian Johner
Sehr geehrter Herr Johner,
Wir sind IT Partner vom einem großen KH und übernehmen die Betriebsführung der Software.
In der MPBetreibVO ist die STK für Produkte nach Anhang 1 gefordert:
1) Produkte zum Monitoring von Vitalparametern
Wie sieht es hier mit Anästhesie-Software und PDMS aus? Diese monitoren ebenfalls Vitalparameter und sind derzeit noch MP Klasse 1.
Wie muss eine STK für diese Produkte, falls erforderlich, aussehen? Wenn der Hersteller die sicherheitsrelevanten Funktionen nicht vorgibt, sind diese anhand dem Stand der Technik durchzuführen. Wo finde ich hier relevante Quellen, was genau bei Software zu prüfen ist?
Sämtliche anderen MPs, die nicht im Anhang I genannt werden, müssen keiner Wartung/Kontrolle unterzogen werden?
Und noch eine Frage bzlg. Einweisung. Ist eine Einweisung der User über ein e-learning/Video Portal gesetzeskonform oder muss diese immer persönlich durch den Hersteller bzw. einer von ihm benannten Person erfolgen?
Herzlichen Dank für die Infos!
Mit besten Grüßen aus Wels
Patricia Peherstorfer
Guten Tag Herr Johner,
wie Sie bereits geschrieben hatten, wird die MPBetreibV ja vom MPG verlangt und stellt im Grunde genommen eine Erweiterung des MPG dar.
Da das MPG durch Inkrafttreten der MDR so nicht mehr existieren wird, frage ich mich ob auch die MDR in ihrem Gesetzestext irgendwo auf eine Betreiberverordnung für Medizinprodukte verweist. Können Sie mir diesbezüglich weiterhelfen?
Mit freundlichen Grüßen
Theresa Reeb
Sehr geehrte Frau Reeb,
das MPG wird es weiter geben. Es muss allerdings an die MDR angepasst werden. Die MPBetreibV wird auch weiter bestehen.
Allerdings hingt das BMG etwas hinterher. Es könnte sein, dass am 20.05.2020 noch nicht alles Gesetze und nationalen Verordnungen angepasst sind.
Viele Grüße, Christian Johner
Sehr geehrtes Johner-Team,
im Bereich „regulatorische Anforderungen an den Betreiber…“ a) wurden die Anpassungen der Paragrafen an die 2017er MPBetreibV noch nicht nachgezogen.
Viele Grüße,
Christian Baudis
Sehr geehrtes Johner Team,
können Sie mir sagen, in §5 MPBetreibV Abs. 1, was eine geignete Ausbildung ist ? Dürfen EUP STK ab aktiven MPG Produkten, trotz einer Schulung durch den Hersteller rechtlich durchführen?
Vielen Dank für Ihre Mühe!
Mit freundlichen Grüßen
Peter Scharbau
Die Person sollte über die i.d.R. technischen Fähigkeiten verfügen, die für die STK notwendig ist. Welche das sind, sollte der Hersteller bestimmen. Falls das nicht fehlt, würde ich das anfragen. Wenn auch dann nichts kommt, müsste eine einschlägige Ausbildung wie zum Medizintechniker ausreichen. Es steht nirgends, dass es der Hersteller selbst sein muss.
Sehr geehrter Herr Prof. Johner!
Können Sie mir mitteilen, wie das bei aktiv implantierten MP nach deutschem Recht ist? Wer ist hier der Betreiber? Die Gesundheitseinrichtung die implantiert hat, oder aber der Anwender, in dessen Besitz das Implantat übergeht? Vielen Dank – Liebe Grüße
Peter Müllner
Lieber Herr Müllner,
es ist die Gesundheitseinrichtung, die implantiert hat. Sie muss nach §13 auch das Bestandsverzeichnis führen.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich habe zwei Fragen zur Auslegung der MPBetreibV.
1. Gilt das MP für orthopädische Hilfsmittel, wie maßgefertigte, orthopädische Schuheinlagen?
2. Was bedeutet konkret § 1 Anwendungsbereich, (2) Diese Verordnung gilt nicht für Medizinprodukte, 3. „die in ausschließlich eigener Verantwortung für persönliche Zwecke erworben und angewendet werden“:
Konkreter Fall: Ich möchte gerne eine Firma gründen, die maßgefertigte, orthopädische Schuheinlagen (gem. Standards aus Hilfsmittelverzeichnis der Produktgruppe: 08 „Einlagen“) an einen Endkunden verkauft. Diese werden von einem zertifizierten Orthopädischen Schuhmachermeister produziert, von dem ich sie abkaufe. Ich bin nicht zertifiziert. Diese verkaufe ich einem Endkunden, der sie selbst zahlt (nicht über die Krankenkasse). Bin ich dann vom MPBetreibV befreit und muss keine Qualifikationen gem. MPBetreibV vorweisen?
Danke für Ihre Unterstützung!
Beste Grüße, A. Wolf
Sehr geehrter Herr Johner,
ich interessiere mich für die Auslegung der MPBetreibV:
1) Gilt das MP für orthopädische Hilfsmittel, wie maßgefertigte, orthopädische Schuheinlagen?
2) Was bedeutet § 1 Anwendungsbereich, (2) Diese Verordnung gilt nicht für Medizinprodukte, 3. „die in ausschließlich eigener Verantwortung für persönliche Zwecke erworben und angewendet werden“: wenn ich maßgefertigte, orthopädische Schuheinlagen an einen Endkunden verkaufe, der diese privat bezahlt (nicht über die Krankenkasse einreicht), bin ich als Verkäufer dann vom MPBetreibV und den entsprechenden Voraussetzungen befreit?
Herzlichen Dank für eine Rückmeldung.
Beste Grüße, A. Wolf
Sehr geehrte Frau Wolf,
die Betreiber-Verordnung gilt nicht für Schuheinlagen, weil genau der von Ihnen zitierte Abschnitt das ausschließt. Sie scheinen die Rolle eines Händlers einzunehmen. Die neue MDR stellt an Händler ebenfalls Anforderungen. Dass eine neue MPBetreibV, die geschrieben werden muss, um die Forderungen der MDR abzubilden, daran etwas ändert, dass Sie nicht unter die Verordnung fallen, glaube ich nicht.
Viele Grüße, Christian Johner
Die Grafik unter Punkt 5 referenziert falsche (alte?) Paragraphen. Die Bildbeschreibung referenziert die richtigen Paragraphen bezüglich STK (§11) und MTK (§14) der MPBetreibV.
Danke, Herr K!
Ich werde das gleich am Wochenende angehen. Danke für den Hinweis!
Viele Grüße, Christian Johner
Hallo Herr Johner ich habe eine Frage zum Paragraphen 5.
5 Besondere Anforderungen
(1) Sofern für eine Tätigkeit nach dieser Verordnung besondere Anforderungen vorausgesetzt werden, darf diese
Tätigkeit nur durchführen, wer
1. hinsichtlich der jeweiligen Tätigkeit über aktuelle Kenntnisse aufgrund einer geeigneten Ausbildung und
einer einschlägigen beruflichen Tätigkeit verfügt,
2. hinsichtlich der fachlichen Beurteilung keiner Weisung unterliegt und
3. über die Mittel, insbesondere Räume, Geräte und sonstige Arbeitsmittel, wie geeignete Mess- und
Prüfeinrichtungen, verfügt, die erforderlich sind, die jeweilige Tätigkeit ordnungsgemäß und nachvollziehbar
durchzuführe. So in Punkt 2 steht das
2. hinsichtlich der fachlichen Beurteilung keiner Weisung unterliegt und!
Meine Frage ist ob ich als Leitung einer AEMP dem Arzt gegenüber keiner Weisung unterliege! Da ich die notwendige Ausbildung/Fachkunde besitze.
Danke für ihre Antwort.
Mfg
Herr Heindl
Sehr geehrter Herr Heindl,
eine spannende Frage!
Die Leitung einer AEMP ist eine Funktionsbeschreibung, keine Kompetenzbeschreibung. Daher fällt die Antwort schwer. Die Anforderung, dass keine Weisung erfolgt, scheint erfüllt zu sein.
Somit wären das gute Schrittte:
Hilft das ein wenig? Wenn nicht gerne nachhaken.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
wir sind ein Leistungserbringer im Rettungsdienst, die DRK Rettungsdienst Bergstraße gGmbH. Unser Einsatzgebiet umfasst einen Flächenkreis, so dass wir verschiedene Rettungswachen im Kreisgebiet unterhalten. Unsere Einsatzfahrzeuge sind mit Medizinprodukten ausgerüstet, einige fallen unter die Pflicht in einem Bestandsverzeichnis gelistet zu sein. Bei Werkstatt- oder Serviceaufenthalten von Fahrzeugen werden teilweise Medizinprodukte (Defibrillator, Beatmungsgerät etc.) in Ersatzfahrzeuge umgeräumt um diese Fahrzeuge dann in Einsatz bringen zu können. Als Qualitätsmanagement-Beauftragter werde ich immer wieder mit der Frage konfrontiert: „In welchem Zeitraum (nach dem Wechsel von Medizinprodukten in ein anders Fahrzeug, welches eventuell auch an einer anderen Wache stationiert ist) ist das Bestandsverzeichnis zu aktualisieren?“ Ich würde mich freuen, wenn Sie mir einen Antwort geben könnten.
Mit freundlichen Grüßen
Thomas Peppler
Sehr geehrter Herr Peppler,
danke für die spannende Frage!
Die MPBetreibV nennt dazu keine Fristen. Damit gewährt sie auch keine Fristen. D.h. das Bestandsverzeichnis sollte immer aktuell sein. Dass dies im Einzelfall schwierig sein kann, kann ich gut nachvollziehen.
Auf das Motto „wo kein Kläger, da kein Richter“ sollte man sich eher nicht zurückziehen und gar die Aktualisierung bis auf die in Anlage II genannten Fristen verzögern.
Mittelfristig hilft wahrscheinlich nur ein Warenwirtschaftssystem, mit dem man (semi-)automatisiert das Bestandsverzeichnis aktualisiert. Bis das implementiert ist, muss das Verzeichnis zumindest so aktuell gehalten werden, dass keine STK/MTK verpasst wird. Denn das ist das Hauptanliegen der MPBetreibV in diesem Kontext.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner.
Meine Frage bezieht sich auf den §11 und damit auch auf die Anlage 1.
Wir verwenden Beatmungsgeräte (damit Anlage1-Geräte) im mobilen Einsatz. Damit die Geräte funktionieren, benötigen diese eine entsprechende Sauerstoffversorgung über Sauerstoffflaschen, Druckminderer und Verbindungsleitungen bis hin zum Beatmungsgerät.
Es steht in der BetreibV :
„Für andere Medizinprodukte sowie Zubehör einschließlich Software oder andere Gegenstände, die der Betreiber mit Medizinprodukten nach Satz 1 verbunden verwendet, gelten die Sätze 1 bis 3 entsprechend.“.
Außer dem Beatmungsgerät selbst, ist keines der anderen Komponenten ein Anlage1-Gerät und für mich, sind die gesamten Komponenten, unabhängig ob Schraub- oder Steckverbindung, miteinander verbunden.
Nun meine Frage: Gelten alle anderen Komponenten, durch den Verbund zum Beatmungsgerät, automatisch als Anlage 1 Geräte und damit auch der §11 oder ist „…verbunden verwendet…“ in diesem Zusammenhang anders zu verstehen.
Viele Grüße
Heiko Graff
Sehr geehrter Herr Graff,
danke für die spannende Frage!
Bei der Entscheidung ob für die „anderen Komponenten“ auch eine STK durchgeführt (bzw. die anderen Sätze des §11 Absatz 1 beachtet) werden müssen, hängt von zwei Punkten ab:
Wenn beides der Fall ist, dann werden diese „Komponenten zwar nicht zum „Anlage-1-Gerät“, aber für diese müssen Sie gemäß §11 (1) Satz 5 ebenfalls die Sätze 1 bis 3 befolgen.
Durch das Verbinden der Komponenten mit dem Beatmungsgerät werden diese nicht selbst zum Anlage-1-Gerät, aber eben sehr ähnlich zu behandeln.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
wir verwenden in unserer Rettungsdienstorganisation einen AED eines namhaften Herstellers im qualifizierten Krankentransportbereich. Der Hersteller des AED stellt bei seiner Einweisung der von uns beauftragten Personen (= vom Betreiber beauftragten Person) ein Zertifikat aus, welches die Einweisung nach § 10 MPBetreibV bescheinigt. Die von uns beauftragten Personen sind also berechtigt, weitere Anwender in die sachgemäße Handhabung und Anwendung des AED einzuweisen. Gleiches handhabt der Hersteller auch so bei der Erstinbetriebnahme.
Nun befindet sich aber auf dem Zertifikat, der die Einweisung bescheinigt, folgender Text: „Herr/Frau Manfred Mustermann wurde nach § 10 MPBetreibV auf den AED XYZ eingewiesen (…) Er/Sie ist berechtigt, bis zum 31.12.2022 weitere Anwender in den AED XYZ einzuweisen“. Der Hersteller sieht hier eine zeitliche Befristung der Gültigkeit der „Herstellereinweisung“ vor. Sprich: Nach dem 31.12.2022 dürfte Herr Mustermann keine weiteren Anwender einweisen. Aus Sicht der MPBetreibV gibt es aber keine Grundlage zur befristeten Gültigkeit einer Einweisung der vom Betreiber beauftragten Personen. Der Hersteller teilte mir auf Anfrage mit, dies wäre so vorgeschrieben, da der Hersteller für die durchgeführte Einweisung an Herr Mustermann hafte(!?). Der Hersteller konnte mir aber das Gesetz nicht nennen, auf das er seine Aussage stützt. Mir wäre außerdem kein Gesetz aus dem Medizinproduktebereich bekannt, das dies vorschreibt wie vom Hersteller genannt.
Auf welcher rechtlichen Grundlage befristet der Hersteller hier die Gültigkeit? Bzw. kann generell eine solche Einweisung nur eine befristete Gültigkeit haben? Bei anderen Herstellern z. B. von EKG/Defibrillator-Kombinationen (Corpuls) reicht eine einzige Einweisung durch den Hersteller aus – diese ist unbefristet gültig.
Aus meiner Sicht reiche eine einmalige Einweisung für die beauftragten Personen auf den AED aus, zumal unser Personal weitaus komplexere Medizingeräte anwendet und die beauftragten Personen auch ausgebildete Notfallsanitäter sind. Ein AED ist aus unserer Sicht für unser Personal kein komplexes Gerät.
Mit besten Grüßen aus Hessen
Hannah-Lisa Wurger
Sehr geehrte Frau Wurger,
danke für die wirklich spannende Frage!
Mir ist keine gesetzliche Grundlage bekannt, die die Dauer der Wirksamkeit einer Einführung begrenzt. Aber der Hersteller kann frei definieren, welche Maßnahmen er für notwendig hält für die sichere Anwendung seines Produkts. Diese Maßnahmen ergeben sich üblicherweise aus der Risikoanalyse.
Allerdings sind die meisten Hersteller vorsichtig dabei mit übertriebenen Schulungsanforderungen, weil damit die Marktakzeptanz sinkt.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Dr. Johner,
danke Ihnen für die schnelle und hilfreiche Antwort.
Mit dem Hersteller habe ich nochmal telefoniert und eine andere Ansprechpartnerin zugewiesen bekommen. Sie teilte mir, nach Rücksprache mit Ihrer Projekt- und Vertriebsleitung, mit, dass die Einweisungen für die Organisationen (Rettungsdienst, Klinik etc.) unbefristet seien. Sobald eine Einweisung der beauftragten Person durch den Hersteller oder dessen befugte Person, im Medizinproduktebuch dokumentiert sei, ist die Einweisung stets unbefristet. Hier spielt es übrigens keine Rolle, ob das Medizinproduktebuch elektronisch oder in Papierform geführt wird.
Der Hersteller empfehle lediglich jährliche Schulungen durchzuführen für die Betreiber von öffentlich zugänglichen AED’s, was lt. der Ansprechpartner ihr Kollege im Gespräch mit mir verwechselt habe.
Beste Grüße,
Hannah-Lisa Wurger
Sehr geehrter Herr Johner,
welche Länder in Europa, neben Austria und Deutschland hat noch eine ähnliche MPBetreibV??
Oder interpretiert jeder die MDD bzw MDR für sich? Wie soll eine Gesundheitseinrichtung und die vielen Anwender z.B. mit eine Einweisung oder ähnliches in der Praxis umgehen? Falls es nur die MDD bzw. MDR in diesen Ländern gibt. Wie funktioniert dort eine Umsetzung?
Danke
Ihr Achim Storm
Sehr geehrter Herr Storm,
beim Betrieb der Medizinprodukte gibt es bein den regulatorischen Vorgaben in der Tat eine größere „Variabilität“. Das liegt daran, dass weder die MDD noch die MDR präzise Vorgaben dazu machen. Deren Anforderungen wenden sich v.a. an die Hersteller, Händler, Bevollmächtigten und Importeure, sprich an die sog. Wirtschaftsakteure. Die Gesundheitseinrichtungen zählen nicht dazu.
Die vergleichsweise wenigen Anforderungen, die die MDD bzw. MDR an Gesundheitseinrichtungen stellen, haben wir Ihnen im Artikel Auswirkungen der MDR und IVDR auf Gesundheitseinrichtungen wie Kliniken und andere Betreiber zusammengefasst.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
seit Geltungsbeginn der MDR verweisen Hersteller häufiger darauf, dass ihre Medizinprodukte nur mit ihrem Originalzubehör betrieben werden dürfen und nicht mit Zubehör von anderen Anbietern und Begründen dies mit der MDR-Zulassung, die nur für die Kombination Medizinprodukt und Originalzubehör gelten würde.
Z.B.: Der Defibrillator-Hersteller sagt, sein Defibrillator darf nur mit seinen Originalelektroden betrieben werden.
Der Hersteller bzw. der Händler des Nicht-Original-Zubehörs sagt seine Elektrode sei für diesen Defibrillator geeignet.
Wenn der Hersteller bzw. der Händler des Nicht-Original-Zubehörs eine Kompatibiltätsbescheinigung ausstellt in der er die Kompatibilität seines Zubehörs mit dem Gerät bescheinigt, darf man dann weiterhin das Gerät mit Nicht-Originalzubehör betreiben oder kann man als Betreiber/Anwender haftbar gemacht werden?
Viele Grüße
Christoph Hargarter
Sehr geehrter Herr Hargarter,
mit welchem Zubehör ein Medizinprodukt verwendet werden darf, zählt zum bestimmungsgemäßen Gebrauch des Produkts. Diesen legt ausschließlich der Hersteller fest. Das war schon vor der MDR so.
Ein Hersteller eines Zubehörs(!) kann nicht entgegen dieser Festlegung des Herstellers des Medizinprodukts die Konformität des Gesamtsystems erklären. Das sagt Artikel 22 explizit („innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen „).
Dass die Hersteller der Medizinprodukte diese Möglichkeit nutzen, um Hersteller von Zubehör auszugrenzen, ist unbestritten.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich schätze diese Seite sehr und scrolle in unregelmäßigen Abständen mal rein. Nun habe ich hier aber folgendes gefunden. Am 13.05.2019 kam die Frage
„Können Sie mir mitteilen, wie das bei aktiv implantierten MP nach deutschem Recht ist? Wer ist hier der Betreiber? Die Gesundheitseinrichtung die implantiert hat, oder aber der Anwender, in dessen Besitz das Implantat übergeht?“
Ihre Antwort lautete
„es ist die Gesundheitseinrichtung, die implantiert hat. Sie muss nach §13 auch das Bestandsverzeichnis führen.“
Das kann ja schon gleich aus mehreren Gründen nicht sein. Es würde u.a. bedeuten, daß z.B. jeder Schrittmacher ins Bestandsverzeichnis aufgenommen werden müßte…
Sehr geehrte/r L. Müller,
Sie scheinen mit meiner Antwort nicht zufrieden zu sein. Ich bin nun ratlos, was Sie von mir erwarten.
Dass die Gesundheitseinrichtungen bei Implantaten der Klasse III ein Verzeichnis führen müssen besagt, Artikel 27(9).
Viele Grüße Christian Johner
Sehr geehrter Herr Johner,
meine Erwartung ist die Korrektur Ihrer o.g. Aussagen 😉
Die MDR habe ich ohne jeden Zweifel in ihrer Gesamtheit noch nicht erfaßt. Allerdings gibt es ja auch die MPBetreibV zur Umsetzung. In § 2 (2) wird als Betreiber definiert, wer für den Betrieb des Medizinprodukts _in_ der Gesundheitseinrichtung verantwortlich ist. Allerspätestens mit Verlassen der Gesundheitseinrichtung wird das Medizinprodukt nicht mehr in ihr betrieben. Die Gesundheitseinrichtung hat allerspätestens dann keine Verfügungsgewalt mehr über das Medizinprodukt.
Auch versorgt die Gesundheitseinrichtung in diesem Fall keinen Patienten im Sinne von § 3 (2).
Der von Ihnen erwähnte MDR Artikel 27 (9) wird in der MPBetreibV vollständig durch § 15 und die Anlage 3 abgedeckt.
Der ursprünglich von Ihnen genannte § 13 MPBetreibV fordert in (1) explizit nur die Aufnahme aktiver _nichtimplantierbarer_ Medizinprodukte ins Bestandsverzeichnis. Implantate sind also davon ausgenommen.
Kurzum: Die Gesundheitseinrichtung ist weder Betreiber aktiver implantierter Medizinprodukte im Sinne von § 2 MPBetreibV noch muß sie aktive implantierte Medizinprodukte im Bestandsverzeichnis im Sinne von § 13 MPBetreibV führen.
Hallo und Guten Tag,
müssen neu bestellte Gerätebeauftragte, um Einweisungen tätigen zu können eine original Einweisung vom Hersteller bekommen, oder reicht es, wenn bestehende Gerätebeauftragte die `neuen Gerätebeauftragten` schulen, damit diese dann Einweisungen machen können? Was sagt der Gesetzgeber?
Würden uns über eine Rückmeldung freuen.
Intensive Grüße
D. Meyer; J. Konert
Meine Kollegin Astrid Schulze hat dazu folgende Antwort gegeben:
Für die in Anlage 1 aufgeführten Medizinprodukte muss der Hersteller involviert sein. Er muss entweder selbst einweisen oder einer anderen Person die Befugnis erteilen. Das kann der Gerätebeauftragte des Betreibers sein. Hat er also die Befugnis des Herstellers für ein in der Anlage 1 aufgeführtes Medizinprodukt, so darf er andere einweisen. Hat er diese nicht, dann darf er nur die Medizinprodukte einweisen, die nicht in Anlage 1 aufgeführt sind.
Die Einweisung in die ordnungsgemäße Handhabung aktiver nichtimplantierbarer Medizinprodukte ist in geeigneter Form zu dokumentieren.
Begründung:
Die MP BetreiberVerordnung schreibt für Medizinprodukte, die nicht in der Anlage 1 aufgeführt werden, nicht vor, wer die Einweisung vorzunehmen hat.
Siehe §4.
Für die in der Anlage 1 aufgeführten Medizinprodukte gilt laut § 10:
(2) In der Anlage 1 aufgeführte Medizinprodukte dürfen nur von Personen angewendet werden, die durch den Hersteller oder durch eine nach Absatz 1 Nr. 2 vom Betreiber beauftragte Person unter Berücksichtigung der Gebrauchsanweisung in die sachgerechte Handhabung dieses Medizinproduktes eingewiesen worden sind.
§10, Absatz 1 Nr. 2 sagt aus, dass der Betreiber ein in der Anlage 1 aufgeführtes Medizinprodukt nur betreiben darf, wenn zuvor der Hersteller oder eine dazu befugte Person, die im Einvernehmen mit dem Hersteller handelt,
1.
…
2.
die vom Betreiber beauftragte Person anhand der Gebrauchsanweisung sowie beigefügter sicherheitsbezogener Informationen und Instandhaltungshinweise in die sachgerechte Handhabung, Anwendung und den Betrieb des Medizinproduktes sowie in die zulässige Verbindung mit anderen Medizinprodukten, Gegenständen und Zubehör eingewiesen hat.
Eine Einweisung nach Nummer 2 ist nicht erforderlich, sofern diese für ein baugleiches Medizinprodukt bereits erfolgt ist.
Ich hoffe, wir konnten Ihnen damit weiterhelfen!
Herzliche Grüße
Anja Segschneider | Redaktion
Guten Tag Allerseits,
ich hätte eine Frage zur Instandhaltung von Medizinprodukten.
Die „neue“ MPBetrV legt die Prüffrist für STK ja in die Hände der Betreiber und kann mit einer entsprechenden Gefährdungsbeurteilung auf max. 24 Mon ausgedehnt werden.
Soweit so gut. Nun steht aber bei Wartung und Instandhaltung: Grundsätzlich sind gemäß §7 MPBetreibV Instandhaltungsmaßnahmen (Inspektion/Prüfung, Wartung) unter Berücksichtigung der Angaben des Herstellers durchzuführen, der diese Angaben dem Medizinprodukt (Gebrauchsanweisung) beizufügen hat.
Bei einigen Geräten gibt es hier jährliche Wartungsfristen vom Hersteller in der Bedienungsanleitung. Kann man nun die Wartungsfrist analog der STK mit entsprechender Begründung (Gefährdungsbeurteilung) auch bspw. auf 24 Mon. verlängern ?
Vielen Dank und viele Grüße
C. Kammerpein
Guten Tag,
Prof. Johner hat mir die folgende Antwort gegeben:
Die MPBetreibV legt für einige in den Anhängen definierte Produktklassen die Anforderungen an die STKs bzw. MTKs fest. Wenn die Produkte nicht in diese Klasse fallen, dann sind die Betreiber „nur“ an die Vorgaben der Hersteller gebunden.
Die Hersteller leiten die Frequenz für die Kontrollen aus dem Risikomanagement ab. Damit ist es i.d.R. für Betreiber schwer, zu einem anderen Ergebnis zu kommen. Das gilt insbesondere, weil die Betreiber die Architektur des Geräts nicht kennen und damit die Einflussfaktoren auf die Frequenz. Eine Ausnahme wären Annahmen, die der Hersteller trifft und kommuniziert, die in der Praxis beim Betreiber nicht gegeben sind. Aber selbst dann würde ich es für sinnvoll halten, eine geänderte Frequenz mit dem Hersteller abzustimmen. Denn die Wartung gehört zum expliziten bestimmungsgemäßen Gebrauch.
Ich hoffe, wir konnten Ihnen damit helfen!
Herzliche Grüße
Anja Segschneider | Redaktion
Hallo,
aus mir nicht erklärlichen Gründen werden Laborgeräte (IVD) kaum in der MPBetreibV erwähnt.
Sie fallen auch nicht unter die Medizinprodukte der Anlage 1&2.
Es wird zwar auf die RiliBÄK verwiesen, die aber manche Aspekte nicht besonders detailliert vorgibt. Mir geht es dabei um die Geräteeinweisungen.
Im Speziellen will ich hier auf die, bei Geräten der Anlage 1&2, vorgeschriebene Einweisung eines Gerätebeauftragten hinaus.
Ich bin überrascht, dass es hier für Laborgeräte keine ähnliche Vorgabe gibt.
Findet man diese in einem anderen Gesetzestext?
Ich komme aus dem Laborbereich und kenne nur das Vorgehen, dass die Gerätebeauftragten vom Hersteller eingewiesen werden, um dann dafür verantwortlich zu sein, dass die anderen Anwender durch die Gerätebeauftragten geschult werden und dass die Geräte nach Herstellervorgabe gepflegt und gewartet werden. Ich hatte immer den Eindruck, dass dies Aufgrund von Gesetzen oder Richtlinien erfolgt, finde aber keinen passenden Gesetzestext.
Viele Grüße,
S.Becker
Sehr geehrte Frau Becker,
vielen Dank für Ihre Frage und bitte entschuldigen Sie die ungewohnt lange Wartezeit.
Prinzipiell versucht die MPBetreibV auf alle Medizinprodukte Rücksicht zu nehmen, die IVD-Laborgeräte nehmen dabei nur eine kleine Untergruppe ein.
Letztlich spezifiziert Teil A der Rili-BÄK detaillierter, welche Anforderungen durch medizinische Labore – Betreiber von IVD – zu erfüllen sind.
Ein Gerätebeauftragter oder auch Geräteverantwortlicher wird durch die Rili-BÄK nicht explizit gefordert. Ein Gerätebeauftragter ergibt sich jedoch indirekt aus der Forderung, dass medizinische Labore „über ein Verfahren für die regelmäßige Überwachung der Funktion der Geräte […] verfügen und dieses umsetzen [müssen]“. Darüber hinaus ist für jedes Gerät eine Dokumentation zu führen, was diese beinhalten soll, spezifiziert die Rili-BÄK und die Geräte „dürfen nur durch hierzu befugte und eingewiesene Mitarbeiter bedient werden“ (Rili-BÄK, Teil A, 5.4).
Außerdem ist es für ein QM-System nicht nur üblich sondern auch gefordert, Verantwortlichkeiten festzulegen, so gibt es auch die Rili-BÄK vor (Rili-BÄK, Teil A, 7.1.1).
Den Forderungen hinsichtlich der Geräte, kommt man am ehesten nach indem man einen Geräteverantwortlichen oder Gerätebeauftragten festlegt, dies ergibt sich auch aus der guten Laborpraxis.
Teil A der Rili-BÄK ist angelehnt an die Forderungen der ISO 15189. Auch in der Norm zum QMS in medizinischen Laboren werden Anforderungen hinsichtlich der „Ausrüstung“ (Geräte inkludiert) festgelegt, dies beinhaltet u. a. Annahmeprüfungen, Kennzeichnung, Instandhaltung und Reparatur, Personalbefugnisse, Meldung von Vorkommnissen und Aufzeichnungen. Ein Großteil der medizinischen Labore ist akkreditiert nach ISO 15189.
Darüber hinaus gibt nicht nur die MPBetreibV vor, dass die Verwendung von IVDs nur im Rahmen geeigneter QM-Systeme gestattet ist, sondern auch die IVDR legt dies auf europäischer Ebene fest [IVDR, Art. 5 (5) b) und c)].
Ich hoffe, meine Ausführungen helfen Ihnen weiter.
Viele Grüße Sophie Bartsch
Guten Tag,
zum Thema MPBetreibV und Softwarehersteller. Sie schreiben „Wann sind Hersteller gleichzeitig Betreiber, fragen Sie sich vielleicht. Bei den Mobile Medical Apps ist das inzwischen eher die Regel als die Ausnahme, denn die Inverkehrbringer, die Medizinproduktehersteller, betreiben einen Server, der Daten und teilweise auch wesentliche Teile der Logik enthält und dessen Software ein Medizinprodukt oder ein Teil eines Medizinprodukts ist.“
In welchen Fällen ist denn denkbar, dass der Hersteller einer „on premises“ installierten Standalone-Software zum Betreiber werden könnte?
Beste Grüße,
M. Heller
Sehr geehrter Heller,
danke für Ihre Frage!
Mir ist keine Situation bekannt, in der ein Hersteller einer „on premise Lösung“ Betreiber wäre. Es sei denn es ist eine Eigenherstellung einer Klinik.
Ich sehe keine Gefahr, dass ein Hersteller die Software im Rahmen der Zweckbestimmung selbst nutzt oder dies versehentlich tun würde.
Viele Grüße
Christian Johner
Hallo liebes Johner Team,
sind Ihnen im internationalen Umfeld Regularien ähnlich der MPBetreibV bekannt? Also wenn ich z.B. in Spanien, Italien oder ganz allgemein Weltweit ein Medizinprodukt betreiben möchte, wo/wie sind dann jeweils meine Pflichten geregelt? Ich denke da z.B. an einen medizinischen Cloud Service (SaaS).
Aus meiner Sicht wäre es naheliegend, wenn in jedem Land in dem ich den medizinischen Service zugänglich mache und aufrecht erhalte eigene nationale Anforderungen an den Betrieb gelten. Bei grob 200 Staaten weltweit wäre das vermutlich ein ziemlich kompliziertes Unterfangen.
Bei der Inverkehrbringung hat man als klassischer Medizinproduktehersteller ja durchaus Erfahrung, aber wenn man plötzlich Betreiber wird…
Schon mal herzlichen Dank für eventuelle Tipps.
Viele Grüße
Claus Leiendecker
Sehr geehrter Herr Leiendecker,
es gibt in den meisten Ländern nationale Gesetze und Verordnungen. Wie sehr die den Betrieb regeln, kann ich pauschal nicht sagen, weil dazu mehrere Rechtsbereiche zu untersuchen sind.
Die Hoheit, die datenschutzrechtlichen Vorgaben zu machen, liegen bei der EU. Die hat nicht nur die DSGVO veröffentlicht, sondern auch besondere Vorgaben wie den Cloud Act. Allerdings sind nicht alle Staaten ganz konsequent bei der Umsetzung des EU-Rahmens. Das ist nicht nur in Deutschland mit seinen vielen Landesdatenschutzbeauftragten der Fall, sondern auch beispielsweise in Frankreich.
Mein Tipp wäre, zum einen mit einem spezialisierten Anwalt (wir können bei der Suche helfen), Land für Land die lokalen Ausprägungen zu recherchieren, zum anderen sicherzustellen, dass die (geplanten) EU-Vorgaben erfüllt werden (können). Wenn ich richtig erinnere, ist bei FMC in den Landesorganisationen viel Wissen vorhanden.
Viele Grüße
Christian Johner
Sehr geehrter Herr Prof. Johner,
vielen Dank für Ihre Einschätzung. Im Grundsatz deckt sich das ja mit meiner Vermutung.
Ich hätte noch eine Frage zu dem von Ihnen erwähnten Cloud Act: Ist das eine EU Vorgabe? Ich finde bei Google nur einen Cloud Act aus den USA.
Viele Grüße und ein schönes Wochenende
Claus Leiendecker
Sehr geehrter Herr Leiendecker,
Sie haben absolut Recht: Ich meinte nicht den EU Cloud Act, sondern den EU Governance und EU Data Act. Beide wirken sich zumindest indirekt auf den Datenschutz aus:
In Gegensatz zur DSGVO geht es aber nicht primär darum, Daten zu schützen, sondern auch darum, Daten nutzbar zu machen. Sie finden in diesem Artikel. Diese Nutzbarmachung hat jedoch wieder datenschutzrechtliche Implikationen. Ein paar Gedanken finden Sie im verlinkten Artikel.
Nochmals danke für Ihren wichtigen Hinweis!
Viele Grüße
Christian Johner
Sehr geehrtes Team,
meine Frage bezieht sich auf Punkt 6, hier schreiben Sie wie folgt:
>>Sicherheitstechnische Kontrollen haben das Ziel, die Sicherheit von Medizinprodukten zu überprüfen und sicherzustellen, um Gefährdungen für Patienten, Anwender und Dritte zu minimieren.
Die STKs sind aber nur für die in Anlage 1 gelisteten Medizinprodukte vorgeschrieben. Das sind im Wesentlichen kritische aktive, nicht implantierbare Produkte wie Kernspingeräte, Stimulatoren, Beatmungsgeräte oder Geräte zur Lithotripsie. Allerdings kann der Hersteller unabhängig von der Anlage I eine STK verlangen, wenn gleich die MPBetreibV in § 7 von Wartung spricht:
„Wartungen, die erforderlich sind, um den sicheren und ordnungsgemäßen Betrieb der Medizinprodukte fortwährend zu gewährleisten. Die Instandhaltungsmaßnahmen sind unter Berücksichtigung der Angaben des Herstellers durchzuführen, der diese Angaben dem Medizinprodukt beizufügen hat.“<<
Das Bundes Gesundheitsministerium schreibt in den FAQ zur MPBetreibV das folgende:
„Sind sicherheitstechnische Kontrollen durchzuführen, bei denen der Hersteller z.B. in der Gebrauchsanweisung eine solche Kontrolle fordert?
Nein, da ausschließlich Produkte der Anlage 1 einer sicherheitstechnischen Kontrolle zu unterziehen sind.
Hinweis: manche Hersteller haben unter dem Begriff „sicherheitstechnische Kontrolle“ die Kontrolle verstanden, die der Hersteller im Rahmen von Instandhaltungen in der Gebrauchsanweisung aufführen muss. Solche Kontrollen sind zu beachten. Davon sind aber die durch den Verordnungsgeber geforderten sicherheitstechnischen Kontrollen nach § 11 strikt zu unterscheiden.“
(QUELLE: https://www.bundesgesundheitsministerium.de/faq-mpbetreibv.html)
Wer hat nun recht?
Wie verhält sich das ganze bei einem Pflegebett?
Der Hersteller fordert eine STK, das BGM sagt nein. – Reicht hier tatsächlich die Prüfung nach DGUV V3?
Mit freundlichem Gruß
Kai Demel
Sehr geehrter Herr Demel,
danke für Ihre wichtige Frage. Der Widerspruch folgt meines Erachtens dadurch, dass der Begriff STK nicht einheitlich verstanden wird. Manche beschränken ihn auf die Definition der MPBetreibV. Andere verstehen darunter Kontrollen, die der Hersteller vorsieht, um die Sicherheit des Medizinprodukts zu prüfen und zu gewährleisten.
Im Fall eines Pflegebetts bedarf es keiner STKs, da sie nicht in der Anlage 1 stehen, wie Sie völlig korrekt anmerken. Wenn der Hersteller aber Kontrollen verlangt, um die Sicherheit seiner Produkte zu gewährleisten, dann sollte man dem folgen. In weit diese Kontrollen sinnvoll sind, kann ich nicht beurteilen. Es gibt Hersteller, die mit den Kontrollen Geld verdienen wollen. Andere glauben irrtümlich, sie müssten diese Kontrollen fordern. Diese Frage würde ich mit dem Hersteller direkt klären.
Herzliche Grüße
Christian Johner
Guten Tag,
Mich beschäftigt in letzer das Thema Prüfung vor Erst-Inbetriebnahme. Die STK ist laut Gesetztestext spätestens nach 24 Monaten fällig. Das ist klar.
Im Gesetzt steht aber: Der Betreiber des Hilfsmittels ist nach MPBetreibV § 4 verpflichtet, bei jedem Neuaufbau, jeder Instandhaltung und im laufenden Betrieb regelmäßige Prüfungen durchzuführen, um den si-
cheren Zustand des Hilfsmittels zu gewährleisten.
Die Prüfung beim Neuaufbau (z.B. Bett) beim Endkunden ist für mich nicht klar geregelt. Muss bei einem Wechsel in einen anderen Raum und vorherigen Abb- und Aufbau des Bettes erneut eine Prüfung erfolgen?
Könnten Sie mir mitteilen, ob die Prüfung vor der Lieferung an den Endkunden vor Ort passieren muss oder kann die Prüfung im Unternehmen vorher stattfinden?
Kann das ggf. sogar von den Vorgaben des Herstellers abhängen?
LG
Sehr geehrter Herr Petker,
STK-Prüfungen sind gemäß § 11 MPBetreibV nur für bestimmte Medizinprodukte erforderlich, die in Anlage 1 der Verordnung aufgeführt sind. Patientenbetten zählen nicht dazu. Dennoch müssen alle medizinischen elektrischen Geräte geprüft werden. Die DGUV Vorschrift 3 (DGUV V3) legt die Prüfpflicht für elektrische Betriebsmittel fest und fordert in § 5 Prüfungen vor der ersten Inbetriebnahme, nach Änderungen oder Instandsetzungen sowie in regelmäßigen Abständen. Die DIN EN 62353 konkretisiert diese Anforderungen speziell für den medizinischen Bereich und sieht Sichtprüfungen, elektrische Messungen und Funktionsprüfungen vor.
Die DIN EN 62353 sieht eine Prüfung vor Inbetriebnahme vor Ort vor. Lediglich wenn vom Hersteller die Ergebnisse der Fertigungsendprüfung in den Begleitpapieren zur Verfügung gestellt werden, können diese die Prüfung vor Ort ersetzen.
Für Wiederholungsprüfungen sind gemäß Anhang F der DIN EN 62353 Prüffristen zwischen 6 und 36 Monaten festzulegen, sofern der Hersteller keine Prüffristen in den Begleitdokumenten vorgibt. Für bestimmte medizinische Geräte sollte eine maximale Frist von 24 Monaten nicht überschritten werden. Die Festlegung der Prüffristen hängt von Faktoren wie der Nutzungshäufigkeit des Geräts, der Betriebsumgebung und der Betriebsweise (stationär, mobil, Notfalleinsatz) ab. Lediglich ein Raumwechsel eines Medizinprodukts erfordert grundsätzlich keine erneute Prüfung. Stattdessen sollte die Prüffrist so festgelegt werden, dass sie der Mobilität des Geräts gerecht wird. Fahrbare Geräte, wie Patientenbetten, die bspw. in einem Krankenhaus häufig zwischen Stationen bewegt werden, könnten aufgrund der regelmäßigen Ortsveränderungen kürzere Prüfintervalle erfordern.
Wenn ein Medizinprodukt bzw. in Ihrem Fall ein Patientenbett ab- und aufgebaut wird, kann eine erneute Prüfung nötig sein, abhängig von Art und Umfang der Änderungen. Gemäß DIN EN 62353 müssen nach Änderungen an Medizinprodukten, die als Veränderung der konstruktiven oder funktionellen Merkmale in einer nach den Begleitpapieren nicht vorgesehenen Form definiert sind, erfolgen. Es ist jedoch in jedem Fall zu empfehlen, nach der Montage eine Sichtprüfung sowie eine Funktionsprüfung durchzuführen, um sicherzustellen, dass keine Schäden vorliegen und das Bett wieder in den für den bestimmungsgemäßen Gebrauch notwendigen Zustand versetzt wurde.
Herzliche Grüße
Ayleen Stade
Einen schönen guten Tag,
in meinem Arbeitsumfeld kursiert seit geraumer Zeit der urbane Mythos, dass Geräte, die nicht der Anlage 1 angehören, jedoch in Kombination mit STK-Pflichtigen Geräten betrieben werden auch einer STK unterzogen werden müssen. Auf eigene Recherche finde Ich hierzu leider keinerlei Grundlage für diese Annahme. Vielleicht kann mir hier ja weitergeholfen werden. Gibt es eine Grundlage für diese Annahme?
VG und herzlichen Dank.
Lieber Herr Ortmann,
vielen Dank für Ihre Frage.
Die MPBetreibV verlangt in § 11 Absatz 1, dass der Betreiber für die in der Anlage 1 aufgeführten Medizinprodukte sicherheitstechnische Kontrollen nach den allgemein anerkannten Regeln der Technik durchzuführen oder durchführen zu lassen hat. In Satz 4 desselben Absatzes wird explizit klargestellt, dass diese Verpflichtung auch für andere Medizinprodukte sowie Zubehör einschließlich Software oder andere Gegenstände gilt, die der Betreiber mit Medizinprodukten aus Anlage 1 verbunden verwendet.
Herzliche Grüße,
Ayleen Stade
Sehr geehrte Frau Stade,
ich hoffe, Sie können mir weiterhelfen. Ich würde gerne einschätzen wollen, inwieweit die Medizinprodukte-Betreiberverordnung (MPBetreibV) und die damit verbundenen Folgegesetze auch in der forensischen DNA-Analyse Anwendung finden müssen.
Da forensische DNA-Analysen nicht in Krankenhäusern, sondern eher in kriminaltechnischen Instituten durchgeführt werden – etwa zur Bestimmung von STR-Profilen oder für Vaterschaftsnachweise – stellt sich mir die Frage, ob und in welchem Umfang die entsprechenden Vorschriften auch hier zu beachten sind.
Ich wäre Ihnen dankbar, wenn Sie mir dazu eine kurze Einschätzung geben könnten.
Mit freundlichen Grüßen und herzlichen Dank
Sehr geehrter Herr Wagner,
vielen Dank für Ihre Frage.
Gemäß der MPBetreibV ist der Begriff der „Gesundheitseinrichtung“ in § 2 Abs. 4 weit gefasst und umfasst nicht nur klassische medizinische Einrichtungen wie Krankenhäuser oder Arztpraxen, sondern auch andere Einrichtungen und Institutionen, in denen Produkte gemäß § 3 Nr. 1 des MPDG betrieben und benutzt werden.
Demnach müssen auch kriminaltechnische Institute oder forensische Labore, die Medizinprodukte bzw. In-vitro-Diagnostika einsetzen, die Anforderungen der MPBetreibV erfüllen und sicherstellen, dass diese eingehalten werden. Dazu gehören unter anderem die allgemeinen Anforderungen zur sicheren Nutzung (§ 4) sowie die Instandhaltung der Produkte (§ 7).
Herzliche Grüße,
Ayleen Stade
Sehr geehrtes Team,
die alten Fassung des MPBetreibV hatte unter §14 Abs. 6 die Messtechnische Kontrollen geregelt (dies ist auch auf den Formblättern der ehemaligen Anzeigen vermerkt).
In der neuen Fassung ist die MTK unter §15 geregelt und Abs. 6 wurde gekürzt und es fehlt der Verweis auf die Anzeige von Mitarbeiter die Messtechnische Kontrollen durchführen bei den zuständigen Behörden.
Die Voraussetzungen bzw. die „Besonderen Anforderungen“ gemäß §5 MPBetreibV wurden ebenfalls gekürzt und Abs. 2, welcher der entscheidende war, wurde entfernt.
Nun meine Frage, ist die Azeige der Mitarbeiter demzufolge nicht mehr notwendig und eine firmeninterne Dokumentation (Qualifikationsnachweis etc.) reicht diesbezüglich aus?
Danke im Voraus.
MFG
Sehr geehrter Herr Schmiedel,
korrekt, die Anzeige der Mitarbeiter, die MTK durchführen ist nicht mehr notwendig.
Der Beschluss des Bundesrates zur Drucksache 251/24 (https://www.bundesrat.de/SharedDocs/drucksachen/2024/0201-0300/251-24(B).pdf) enthält dazu auf S. 10 folgende Begründung: „Da die […] vorgeschriebene Anzeige nur den Zustand zum Zeitpunkt vor der Aufnahme der Tätigkeit abbildet und sich die Identifikation der entsprechend beauftragten Personen und Dienste, die messtechnische Kontrollen durchführen aus der jeweiligen Dokumentation ergibt, beinhaltet die Anzeige keinen zusätzlichen dauerhaften Nutzen. Die Streichung der bereits bestehenden Anzeigepflicht im Zusammenhang mit der Durchführung messtechnischer Kontrollen dient der Anpassung an die Regelungen für sicherheitstechnische Kontrollen und zugleich der Entbürokratisierung.“
Herzliche Grüße,
Ayleen Stade
Sehr geehrte Frau Stade,
Der §12 wurde unter dem Abschnitt (3) ergänzt:
„2. das Produkt nach den erfolgreichen sicherheitstechnischen Kontrollen mit einem Zeichen zu kennzeichnen, aus dem das Jahr und der Monat der nächsten sicherheitstechnischen Kontrolle und die Person, die die sicherheitstechnische Kontrolle durchgeführt hat, eindeutig und rückverfolgbar hervorgehen.“
Wie ist hier die Kennzeichnung auf dem Produkt mit der Person, welche die sicherheitstechnische Kontrolle durchgeführt hat „eindeutig und rückverfolgbar hervorgehen“ zu verstehen?
Muss die Person mit seinem Klarnamen (Vor- und Nachname) das Produkt „kennzeichnen“ oder würde hier eine Nummer ausreichen, die dann auch im Protokoll zusammen mit dem Klarnamen steht.
Diese Anforderung stand nicht im letzten Entwurf der Betreiberverordnung, wodurch ich erst jetzt damit konfrontiert worden bin.
Viele Grüße
Ralf Mack
Sehr geehrter Herr Mack,
vielen Dank für Ihre Frage.
Sie haben Recht, diese Anforderung war im Entwurf der MPBetreibV, der im Mai 2024 dem Bundesrat vorgelegt wurde (Drucksache 251/24), nicht enthalten. Sie wurde im Beschluss des Bundesrates im Juli 2024 ergänzt (Beschlussdrucksache 251/24(B)), siehe S. 8.
Zur Rückverfolgbarkeit der prüfenden Person:
Die Formulierung „eindeutig und rückverfolgbar hervorgehen“ bedeutet nicht zwangsläufig, dass der Name der Person direkt auf der Plakette stehen muss.
Eine eindeutige Prüfnummer oder ein Prüfer-Kürzel, das im Prüfprotokoll dem Klarnamen zugeordnet ist, dürfte ausreichen, sofern die Identität des Prüfers zweifelsfrei ermittelbar bleibt. Entscheidend ist, dass die Kennzeichnung auf dem Produkt eine einfache und schnelle Rückverfolgbarkeit ermöglicht, sodass die Person bei Bedarf und ohne großen Aufwand eindeutig identifiziert werden kann.
Herzliche Grüße,
Ayleen Stade
Sehr geehrte Frau Stade,
diesbezüglich haben wir wegen dieser „eindeutig und rückverfolgbar hervorgehen“ ein sehr großes Diskussionspotenzial. Ich glaube nicht, dass jeder den entsprechenden Prüfer, auf die Prüfplatte namentlich oder Prüfer-Kürzel, sofern man keine eigene Prüfplaketten erstellt, „quetschen“ kann.
In unserem Unternehmen laufen im Monat durchschnittlich zwischen 400 und 600 STK durch.
Jedes Medizinprodukt, welches bei uns einer STK durchgeführt wird, wird entsprechend mit einer „vorgefertigten“ Prüfplakette (auf dem man den nächsten STK-Termin per Stanzen herauslesen kann) und einem Protokoll versehen, welches der Betreiber mit seinem Produkt erhält. Auf Grund des Durchlaufes ist zur Zeit nicht anders möglich.
Da die Prüfplakette „relativ“ klein ist, gibt es keinen Spielraum um irgendwelchen Prüfer-Kürzel hinzu zufügen. Auf dem beigefügten Protokoll „eindeutig und rückverfolgbar ist die Person, welche die sicherheitstechnische Kontrolle durchgeführt hat „eindeutig und rückverfolgbar hervorgehbar“ .
Deswegen ergibt sich für mich in diesem Fall kein Sinn, die Prüfer/Durchführer darauf festzunageln.