
Orphan Medical Devices sind Medizinprodukte (und IVD) für kleine Patientengruppen. Die Entwicklung dieser Nischenprodukte rechnet sich für die Hersteller oft nicht. Das führt dazu, dass besonders vulnerable Gruppen wie Kinder nicht immer ausreichend medizinisch versorgt werden können.
Dieser Artikel zeigt Lösungsansätze auf und ordnet die Leitlinie MDCG 2024-10 ein.
1. Orphan Medical Device: Definition und Beispiele
1.1 Allgemeine Definition
Der Begriff Orphan Medical Device (manchmal auch Orphan Device) wurde von den „Orphan Drugs“ auf Medizinprodukte übertragen.
Der Begriff „Orphan Drugs“ (Orphan-Arzneimittel) bezeichnet Arzneimittel für die Behandlung seltener Krankheiten. Abhängig von der Legislative liegen die Kriterien für diese Einstufung zwischen einem und acht Patienten pro 10.000 Einwohner. Entsprechend lassen sich Orphan Medical Devices definieren.
Ein Medizinprodukt oder IVD, dessen Zweckbestimmung sich speziell an kleine Patientengruppen wendet, wobei mit „klein“ nicht mehr als (beispielsweise) 0,5 Promille der Bevölkerung gemeint ist.
Zweckbestimmungen, die auf diese kleinen Patientengruppen zielen, werden auch als Orphan Indication bezeichnet und die Patientengruppen als Orphan Population.
1.2 Definition der MDCG
In der Leitlinie MDCG 2024-10 verwendet die Medical Device Coordination Group (MDCG) eine eigene Definition.
the device is specifically intended to benefit patients in the treatment, diagnosis, or prevention of a disease or condition that presents in not more than 12,000 individuals in the European Union per year; and at least one of the following criteria are met:
- there is insufficiency of available alternative options for the treatment, diagnosis, or prevention of this disease/condition, or
- the device will offer an option that will provide an expected clinical benefit compared to available alternatives or state of the art for the treatment, diagnosis, or prevention of this disease/condition, taking into account both device and patient population-specific factors.
Für die MDCG zählt ein Medizinprodukt also nur dann als Orphan Device, wenn es nicht nur für eine sehr kleine Patientengruppe bestimmt ist, sondern zusätzlich ein Verzicht darauf die Versorgung dieser Orphan Population verschlechtern würde.
1.3 Festlegung der Grenzwerte
Die MDCG legt die Grenze auf 12.000 Patienten pro Jahr in der EU fest. Das entspricht (bei 450 Mio. Einwohnern) etwa 0,27 Promille.
Dieser Wert ist niedrig im Vergleich zu anderen Festlegungen:
- In den USA betreffen Orphan Drugs maximal 0,75 Promille der Bevölkerung (fast das Dreifache).
- Die EU legt in der Verordnung über „Arzneimittel für seltene Leiden“ den Wert auf fünf von 10.000 Personen fest, also 0,5 Promille (d. h. fast das Doppelte).
Es ist nicht allgemein bekannt, was die MDCG bewogen hat, diesen Wert so niedrig anzusetzen.
Ob die Anzahl der Patienten so niedrig ist, weil die Krankheit so selten oder weil die Population so klein ist (z. B. nur Frühgeborene), ist für die Definition nicht relevant.
1.4 Beispiele
Ein Beispiel für ein Orphan Device ist ein Herzklappenimplantat, das für die Behandlung einer seltenen Teilpopulation von Patienten mit Herzklappenerkrankungen vorgesehen ist, die spezifische anatomische Merkmale aufweisen, wie eine extreme Dilatation des ventrikulären Ausflusstrakts.
Andere Beispiele umfassen Geräte zur Behandlung einer seltenen Teilpopulation eines ansonsten nicht seltenen Zustands, die eine definitive Intervention in der Neonatalzeit erfordert (z. B. eine seltene Teilpopulation von hämodynamisch signifikantem persistierendem Ductus arteriosus, die eine akute chirurgische Schließung erfordert).
2. Problemstellung
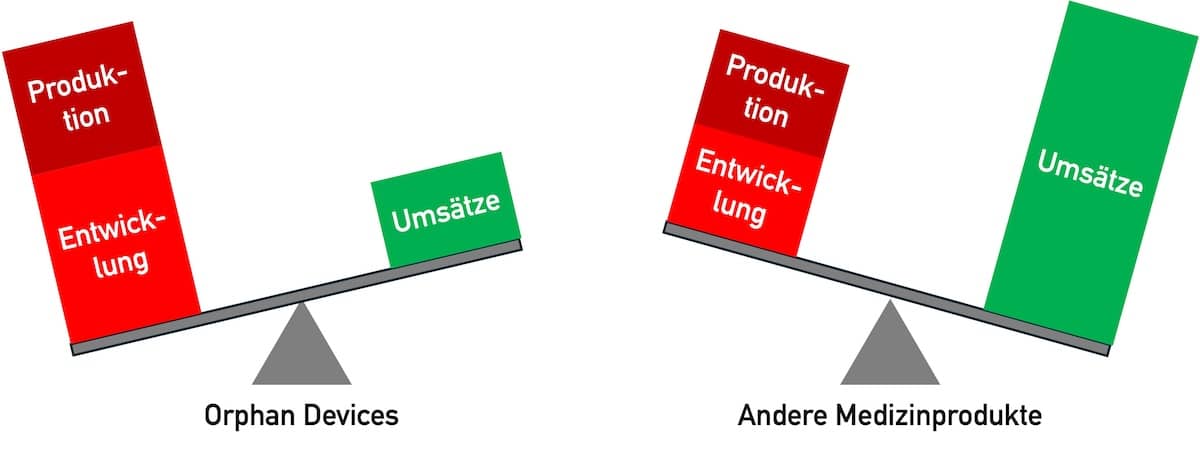
Die Entwicklung von Medizinprodukten ist teuer. Sie rechnet sich bei Orphan Devices aus mehreren Gründen besonders schlecht.
2.1 Sehr hohe Kosten für die Entwicklung von Orphan Devices
Die Kosten für die Entwicklung von Orphan Medical Devices sind aus mehreren Gründen besonders hoch:
- Die Entwicklung ist sehr anspruchsvoll. So sind etwa kleine Ausführungen von Produkten, die dann für Kinder bestimmt sind, nicht mit den gleichen Materialien oder Materialstärken realisierbar.
- Seltene Krankheiten lassen sich oft schwerer diagnostizieren und therapieren. Die Aufwände für die Forschung sind entsprechend hoch, die Entwicklungsrisiken ebenfalls.
- Die klinische Bewertung ist teuer, weil es besonders lange dauert, die für den Evidenznachweis notwendige Fallzahl zu erreichen. Die Rekrutierung der Patienten ist zeit- und kostenaufwändiger als bei einem Produkt für große Patientengruppen.
2.2 Sehr hohe Produkt(ions)stückkosten
Produkte für wenige Patienten werden nur in geringen Stückzahlen gefertigt. Daher profitieren die Hersteller nicht von den Skalierungseffekten, wie sie in einem Massenmarkt auftreten.
2.3 Niedrige Umsätze
Produkte für seltene Krankheiten werden selten benötigt. Entsprechend niedrig sind die erwartbaren Umsätze.
2.4 Zwischenfazit
Den hohen Kosten für Entwicklung und Produktion stehen niedrige Umsätze gegenüber. Diesen Spagat können Hersteller meistens nicht durch höhere Verkaufskosten ausgleichen. Daher bieten sie für Orphan Populations keine Produkte (mehr) an. Eine unzureichende medizinische Versorgung kann die Folge sein.
Diese Unterversorgung betrifft diese Patienten gleich doppelt:
- Die Hersteller entwickeln keine Medizinprodukte und IVD speziell für diese Patientengruppe.
- Die Hersteller schließen Orphan Indications aus der Zweckbestimmung für breitere Patientengruppen aus, weil die Kosten für die spezifischen klinischen Nachweise zu hoch wären.
3. Lösungsansätze
3.1 Regulatorische Lösungen
Der regulatorisch „saubere“ Weg bestünde in speziellen Zulassungsverfahren für diese Patientengruppen. Dabei könnte man sich an der Humanitarian Device Excemption (HDE) der FDA orientieren. Diese orientiert sich am „Orphan Drug Act“ und erlaubt Vereinfachungen bei der Zulassung von Produkten für seltene Krankheiten.
Alternativ sind Vereinbarungen und Leitlinien denkbar, die
- explizit oder implizit die gesetzlichen Anforderungen abschwächen,
- Ausnahmen gestatten (z. B. für Produkte, die im gleichen oder in anderen Märkten bereits in den Verkehr gebracht wurden) oder
- Abweichungen von bestimmten gesetzlichen Anforderungen von der (Straf-)Verfolgung ausnehmen.
Den Ansatz der MDCG beschreibt dieser Artikel im Teilabschnitt 3.3).
3.2 Förderungen
Die Entwicklung und Vermarktung von Medizinprodukten und IVD sollte für deren Hersteller auch ökonomisch Sinn ergeben. Der Gesetzgeber hat die Möglichkeit, in diesen Markt nicht nur regulatorisch einzugreifen, sondern auch über Förderungen:
- Subventionierung der Geräte bzw. ihrer Nutzung über die Krankenkassen oder besondere Steuererleichterungen
- Finanzierung erfolgsversprechender Entwicklungen von Produkten für seltene Krankheiten über Ausschreibungen
- Unterstützung der Forschung an Universitäten und Kliniken, um so den Herstellern Kosten für die Forschung und Grundlagenentwicklung teilweise zu ersparen
3.3 MDCG 2024-10
Die EU erkennt (inzwischen) an, dass die deutlich gestiegenen Anforderungen der MDR insbesondere an die klinische Bewertung eine große Herausforderung für die Hersteller von Nischenprodukten darstellen.
3.3.1 Anwendungsbereich
Die Leitlinie MDCG 2024-10 wendet sich an die Hersteller von Nischenprodukten sowie die verantwortlichen Behörden und Benannten Stellen. Die Leitlinie betrifft die MDR, nicht die IVDR.
Zudem müssen die Produkte der im Abschnitt 1.2 vorgestellten Definition genügen, um als Orphan Device zu gelten. Hersteller müssen wissenschaftlich nachweisen, dass diese Voraussetzungen erfüllt sind.
3.3.2 Lösungsansätze
Die MDCG möchte den Akteuren Hilfestellung bei der klinischen Bewertung geben.
- Sie weist auf erlaubte Einschränkungen bei den klinischen Daten hin, die vor der Inverkehrbringung erhoben werden.
- Insbesondere betrachtet sie Extrapolationen von Daten anderer Populationen unter Umständen als akzeptabel, beispielsweise wenn sie über Post-Market-Aktivitäten (PMCF) ausgeglichen werden können.
- Auch auf andere Datenquellen wie von Äquivalenzprodukten, Registern oder durch Off-Label-Use macht sie aufmerksam.
Zum anderen möchte sie den Prozess der „Zulassung“ unterstützen, indem sie
- Benannten Stellen Hilfestellungen bei der Bewertung gibt (z. B. „Certificates with Conditions“) und
- die Rollen von Experts Panels beschreibt. Dabei bleibt aber unklar, wie die Hersteller davon profitieren.
3.3.3 Bewertung der Leitlinie
Das Positive zuerst: Es wurde erkannt, dass Handlungsbedarf besteht, um die medizinische Versorgung von Orphan Populations nicht noch weiter zu behindern.
Die Autoren der Leitlinie MDCG 2024-10 standen vor der eigentlich unlösbaren Aufgabe, einerseits die durch die MDR behinderte Entwicklung und Zulassung von Orphan Devices zu vereinfachen und andererseits die Regeln der MDR nicht auszuhebeln. Denn dazu sind sie nicht befugt.
Das Ergebnis besteht v. a. aus Hilfestellungen wie Erläuterungen und Argumentationshilfen, wie Freiräume genutzt werden können, welche die MDR bereits gibt – aber beispielsweise von Benannten Stellen nicht immer genutzt werden.
Damit besteht der Mehrwert der Leitlinie v. a. darin, dass man sich auf sie berufen kann, wenn man eine überstrenge Auslegung der MDR vermeiden will.
Dass die MDCG 2024-10 an vielen entscheidenden Stellen selbst wieder vage bleibt oder Selbstverständlichkeiten beschreibt, schränkt den Nutzen ein. Ein Lektorat hätte möglicherweise die Qualität erhöht.
4. Zusammenfassung und Fazit
4.1 Ohne Orphan Medical Devices gibt es ein Problem
Für Hersteller ist es wenig attraktiv, Medizinprodukte für sehr kleine Patientengruppen zu entwickeln. Das trifft besonders zu, wenn Gesetze wie die MDR die Aufwände dafür weiter erhöhen.
Das hat zur Folge, dass die EU für diese Patientengruppen ihre eigenen Ziele durchkreuzt, nämlich mit der MDR einen Beitrag zur Gesundheitsversorgung mit sicheren, leistungsfähigen und wirksamen Produkten zu leisten.
4.2 Das lässt sich durch Leitfäden kaum lösen
Leitfäden wie die MDCG 2024-10 können das nur bedingt auffangen, denn sie dürfen gesetzlichen Anforderungen nicht widersprechen. Daher sind Aussagen, dass der Leitfaden Einschränkungen bei klinischen Daten bei Orphan Devices zulassen würde, zumindest missverständlich.
Vielmehr zeigt der Leitfaden die bestehenden(!) Freiheitsgrade auf. Nicht mehr und nicht weniger. Das Problem ist damit weiterhin nicht beseitigt.
4.3 Daher sind andere Ansätze notwendig
Wenn man „Minderheiten“ wie den „Orphan Populations“ eine gleichwertige Gesundheitsversorgung zugutekommen lassen will, muss man mehr tun:
- Gesetze schreiben, die geeignet sind, diese Ziele explizit zu erreichen
- Zu einem risikobasierten Denken zurückkehren
(Risiken minimiert man nicht dadurch, dass man Medizinprodukte vom Markt nimmt; denn dann entsteht das Risiko der „Nichtversorgung“.) - (Förder-)Gelder bereitstellen, um die Versorgung dieser und künftiger Patientengruppen mit seltenen Krankheiten sicherzustellen (das bedingt auch Investitionen in Innovationen für künftige bzw. bisher nicht entdeckte Krankheiten)
4.3 Fazit
Der Umgang mit besonders vulnerablen Gruppen wie Kindern oder Patienten und Patientinnen mit seltenen Krankheiten macht das Problem deutlich, dass Gesetze für diese Gruppen negative Auswirkungen haben können.
Diese versucht man zu mildern durch Leitlinien oder durch neue Gesetze, z. B. die Meldepflicht für nicht (mehr) verfügbare Produkte. Doch es besteht die Gefahr, dass diese Versuche wirkungslos bleiben oder Folgen zweiter Ordnung haben.
Kurz- und langfristige Maßnahmen können Abhilfe schaffen:
| Kurzfristige Maßnahmen | Langfristige Maßnahmen |
| Förderprogramm aufsetzen Für Orphan Medical Devices in der MDR / IVDR risikobasiert Ausnahmen erlauben, z. B. für Legacy Produkte oder Produkte, die im Ausland „zugelassen“ wurden | Proaktives Monitoring von Versorgungslücken etablieren Ziele der Gesetzgebung definieren und priorisieren Regulatory Science etablieren, z. B. um Systeme zu modellieren Mit diesen Erkenntnissen Gesetze anpassen (z. B. die MDR um neue Zulassungsverfahren ergänzen) |
Das Johner Institut unterstützt bereits Gesetzgeber und Behörden außerhalb der EU bei diesen Aktivitäten.


Sehr geehrte Frau Dr Martin,
Ich habe Ihren Beitrag aufmerksam gelesen.
Ich finde Ihre folgende Ausführung anmaßend und bestürzend zugleich: „Dass die MDCG 2024-10 an vielen entscheidenden Stellen selbst wieder vage bleibt oder Selbstverständlichkeiten beschreibt, schränkt den Nutzen ein. Ein Lektorat hätte möglicherweise die Qualität erhöht.“
Korrigieren Sie mich bitte, aber ich kann mich an Ihren Namen bei den Task Force Meetings zu dieser Guidance, welche jeden Freitag für 2 Stunden über Monate hinweg stattfanden, nicht erinnern.
Ich denke, es wäre angemessen, den vielen Experten aus der EU Kommission, der Industrie, der Wissenschaft und der direkten Patientenversorgung einen angemessenen Respekt zu erbringen – zumindest würde ich das von einem so renommierten Unternehmen wie Johner erwarten. Ihre Phrase erinnert wieder einmal mehr an den wachsenden Populismus.
Und letztendlich, Frau Dr Martin, hilft Ihr Kommentar keinem Hersteller weiter, einen gangbaren Weg zu beschreiten.
Mit freundlichen Grüßen
René Bombien
Sehr geehrter Dr. Bombien,
vielen Dank für Ihre Rückmeldung zu meinem Artikel. Ich verstehe, dass Sie als Teil des Gremiums eine andere Perspektive auf die Entstehung der MDCG 2024-10 haben. Dennoch halte ich es für essenziell, regulatorische Vorgaben kritisch zu hinterfragen – nicht aus Missachtung der geleisteten Arbeit, sondern im Interesse der Hersteller. Ich begrüße hier eine sachliche Diskussion.
Unsere Kunden bitten uns regelmäßig um Einschätzungen zu regulatorischen Vorgaben, und es gehört zu unserer Aufgabe, diese nicht nur zu erläutern, sondern auch ihre praktische Anwendbarkeit zu bewerten. In diesem Kontext stelle ich fest, dass die MDCG 2024-10 an vielen Stellen vage bleibt und vor allem bestehende Freiheitsgrade beschreibt, anstatt neue, konkrete Erleichterungen für Hersteller von Orphan Medical Devices zu schaffen.
Einige zentrale Punkte der MDCG 2024-10 und deren Einordnung:
– Definition von Orphan Devices: Ok, endlich mal eine spezifische Anforderung.
– Erleichterungen bei präklinischen Daten: Das ist nichts Neues – in der klinischen Bewertung der Leistungsbewertung kann das ebenfalls akzeptabel sein.
– Nutzung nicht-klinischer Daten: Das ist auch nichts Neues, siehe Leistungsbewertung nach MDR, Artikel 61.10.
– Flexible Ansätze zur Datengenerierung: Äquivalenzbetrachtung ist auch für andere Produkte möglich.
Diese Punkte zeigen, dass die MDCG 2024-10 zwar Anhaltspunkte gibt, aber in vielen Bereichen keine neuen Lösungen liefert, sondern lediglich bestehende Möglichkeiten zusammenfasst. Der praktische Nutzen bleibt daher fraglich.
Darüber hinaus hat mir ein Kollege mit ähnlichem Hintergrund wie Sie, Kinderchirurg in Lübeck, folgendes Feedback gegeben: „Mit großem Interesse haben wir Ihre Einschätzung zu Orphan Devices und zur MDCG 2024-10 gelesen. In der Tat kommen Sie m. E. zu den richtigen Schlussfolgerungen.“ Sein Feedback unterstreicht, dass die Einschätzung zur MDCG 2024-10 nicht nur meiner persönlichen Meinung entspricht, sondern auch von Experten aus der klinischen Praxis geteilt wird.
Ich lade Sie daher ein, die inhaltlichen Punkte meines Artikels sachlich zu diskutieren. Denn im Mittelpunkt sollte stehen, wie wir gemeinsam die Versorgung von Patienten mit seltenen Erkrankungen verbessern können.
Mit freundlichen Grüßen
Dr. Bettina Martin