
PMCF-Studien sind Studien, die Hersteller im Rahmen der klinischen Nachbeobachtung nach dem Inverkehrbringen (Post-Market Clinical Follow-up) verwenden, um fortlaufend die Konformität ihrer Medizinprodukte nachzuweisen.
Hersteller müssen nicht immer Studien durchführen, um die Anforderungen an den PMCF zu erfüllen. Und nicht alle Typen an PMCF-Studien unterliegen den Anforderungen der MDR.
Dieser Artikel fasst die regulatorischen Anforderungen im Kontext von PMCF-Studien kompakt zusammen und gibt Tipps, wie Sie Rechtssicherheit erlangen und unnötige Aufwände und Umwege bei der PMCF vermeiden können.
1. PMCF-Studien: Die Grundlagen
a) Definition
PMCF-Studien sind klinische Prüfungen im Sinne des Artikels 74 MDR, die nach der Inverkehrbringung des Produkts durchgeführt werden. Zu diesem Zeitpunkt trägt das Produkt bereits eine CE-Kennzeichnung.
Eine klinische Prüfung definiert die MDR als „systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird;“
Die Art der klinischen Prüfung bestimmt die regulatorischen Anforderungen. Diese stellt das zweite Kapitel vor.
b) Ziele von PMCF-Studien
Sicherheit und Leistungsfähigkeit von Medizinprodukten nachweisen
Das Ziel der klinischen Nachbeobachtung nach der Inverkehrbringung (der Post-Market Clinical Follow-up) eines Produkts besteht darin, nach dem Markteintritt zusätzliche klinische Daten zum eigenen Produkt zu sammeln und damit die Sicherheit, Leistungsfähigkeit und Wirksamkeit des Produkts fortlaufend zu bewerten.
Mit PMCF-Studien sollen Fragen beantwortet werden wie:
- Sind die Annahmen an die Lebensdauer des Medizinprodukts noch korrekt?
- Gibt es (neue) Risiken, die bisher nicht identifiziert oder/und in der Risikomanagementakte bereits akzeptiert wurden?
- Gibt es einen Off-Label Use des Produkts?
- Erreicht das Produkt tatsächlich den erwarteten Nutzen?
- Ist das Risiko-Nutzen-Verhältnis weiterhin akzeptabel?
Regulatorische Anforderungen erfüllen
Gesetze wie die MDR verpflichten die Hersteller zur proaktiven klinischen Nachbeobachtung. Daher ist für die Hersteller die gesetzliche Pflicht eine weitere Motivation.
Welche Anforderung die Hersteller im Detail zu erfüllen haben, beschreibt das zweite Kapitel.
Die Anforderungen an PMCF-Studien und der Umgang mit ihnen ist ähnlich wie bei den In-vitro-Diagnostika. Nähere Informationen finden Sie dazu in unserem Fachartikel zum PMS.
2. Regulatorische Anforderungen an PMCF-Studien
a) Auf EU-Ebene
Artikel 61 (11) der MDR schreibt, dass die klinische Bewertung und die dazugehörigen Unterlagen während des gesamten Lebenszyklus des Medizinprodukts anhand von klinischen Daten aktualisiert werden müssen. Diese Durchführung soll im Plan für die klinische Nachbeobachtung nach dem Inverkehrbringen (PMCF-Plan) des Herstellers gemäß Anhang XIV Teil B und dem Plan zur Überwachung nach dem Inverkehrbringen (PMS-Plan) gemäß Artikel 84 dokumentiert werden.
Je nach Studiendesign und Intention der Studie gelten hierbei unterschiedliche Regularien. Man unterscheidet im Wesentlichen drei grundlegende Studientypen für PMCF (Abb. 1).
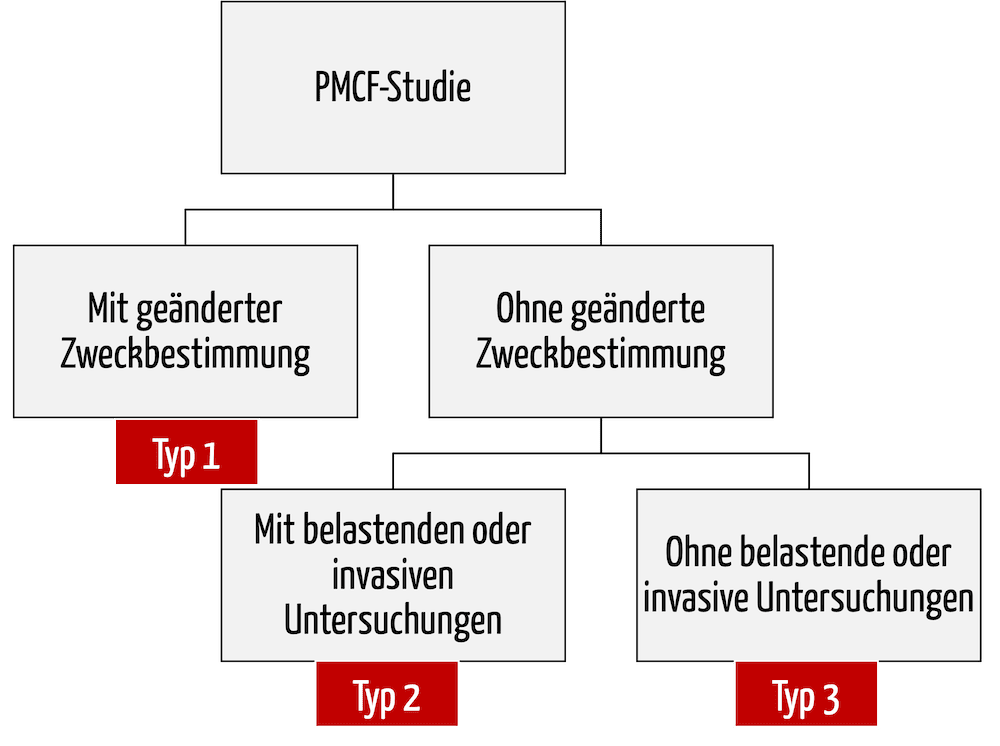
Typ 1: Anwendung des Produkts außerhalb der Zweckbestimmung
Medizinprodukte, die im Rahmen der klinischen Prüfung außerhalb der in der CE-Konformitätserklärung hinterlegten Zweckbestimmung angewendet werden sollen, werden gemäß Artikel 74 (2) reguliert.
Dies wäre der Fall bei einer Erweiterung der Indikation eines Medizinprodukts oder einer Erweiterung der Patientenpopulation.
Somit gelten laut Artikel 74 (2) die Auflagen der Artikel 62-81 MDR und des Anhang XV vollumfänglich (Anforderungen einer Zulassungsstudie nach Artikel 62 zum Nachweis der Sicherheit, Leistung und des klinischen Nutzens).
Falls die Studie in Deutschland stattfindet, müssen die Verantwortlichen auch das MPDG §§ 24 bis 30 und 62 bis 70 berücksichtigen. Für solche Studien erfolgt die Anzeige über das DMIDS (früher DIMDI) bei der lokal zuständigen Ethikkommission. Diese werden von der Bundesoberbehörde (BfArM) geprüft. Das BfArM hat für Sponsoren eine Anleitung hinterlegt, wie klinische Prüfungen im DIMDS zu erfassen sind.
Wichtige Tipps für diese klinischen Prüfungen erhalten Sie in diesem Fachartikel.
Andernfalls gelten die nationalen Gesetze des Landes, in dem die Studienzentren ansässig sind.
Typ 2: Innerhalb der Zweckbestimmung, aber mit belastenden Untersuchungen
Medizinprodukte, die im Rahmen der klinischen Prüfung innerhalb der Zweckbestimmung eingesetzt werden sollen, aber studienbedingt mit zusätzlichen belastenden oder invasiven Untersuchungen geplant sind, werden nach Artikel 74(1) MDR reguliert. Darunter fallen Verfahren, die über die bei normalen Verwendungsbedingungen des Produkts durchgeführten Verfahren hinausgehen und studienbedingt erfolgen.
Die Leitlinie MDCG 2021-6 nennt bei Frage 9 Beispiele für Verfahren, die als „belastend“ oder „invasiv“ eingestuft werden. Eine Blutabnahme oder die Untersuchung durch eine Körperöffnung gelten als invasiv.
Die MDCG schreibt weiter:
„Additional procedures which are burdensome can include a wide variety of different interventions, this may include procedures which may cause pain, discomfort, fear, potential risks or complications/side-effects, disturbances of lives and personal activities, or otherwise unpleasant experiences. It is mostly determined from the perspective of the person bearing the burden.
MDCG 2021-6
Diese Studien müssen die Anforderungen der Artikel 62 MDR Abs. 4 b bis k, m, 75 bis 77, 80(5) und Anhang XI sowie Inhalte des MPDG implizit erfüllen. Gemäß dem deutschen MPDG § 85 Absatz 2 Nr. 6 handelt sich es hierbei auch um ein genehmigungspflichtiges Anzeigeverfahren bei der Bundesoberbehörde, das vom Sponsor über das in Artikel 73 genannte elektronische System mitgeteilt wird (DMIDS), da das in Artikel 78 der MDR erwähnte Verfahren über die EUDAMED noch nicht zur Verfügung steht.
Durch die Bundesoberbehörde erfolgt eine Entgegennahme der Anzeige der klinischen Prüfung. Die Anzeige hat vom Sponsor mindestens 30 Tage vor Beginn der klinischen Prüfung zu erfolgen.
Nähere Informationen des BfArM zu den gesetzlichen Voraussetzungen und weitere Informationen für PMCF-Studien finden Sie auf der Webseite des BfArM.
Typ 3: Innerhalb der Zweckbestimmung und ohne belastende Untersuchungen
Ein Großteil der PMCF-Studien findet innerhalb der Zweckbestimmung und ohne zusätzlich belastende und invasive Untersuchungen statt. In diesen Studien findet die MDR keine Anwendung, sondern es gelten die nationalen Vorschriften und Bestimmungen.
In Deutschland ist das eine optionale berufsrechtliche Beratung gemäß § 15 Berufsordnung der Ärzte. Die Studie muss bei der lokalen Ethikkommission angezeigt werden.
Beachten Sie dabei den Fachartikel zu PMCF und der Notwendigkeit, die Ethikkommission einzubeziehen.
Diese Studien werden in Deutschland auch BO-Ä-Studien genannt. Was Sie beachten müssen und welche Antragsunterlagen Sie brauchen, erfahren Sie in dem Leitfaden „Regulatorische Einordnung von klinischen Prüfungen mit Medizinprodukten“ des Bundesministeriums für Forschung und Bildung.
Zusammenfassung
| Medizinprodukt | Ohne CE | Mit CE (nach Inverkerbringen) | ||
| Anwendung | Vor Inverkehrbringen | Außerhalb der Zweckbestimmung | Innerhalb der Zweckbestimmung | |
| Mit zusätzlichen und belastenden Untersuchungen | Ohne zusätzliche und belastende Untersuchungen | |||
| Typ | Typ 1 | Typ 2 | Typ 3 | |
| Spezifische Anforderungen der MDR | MDR Art. 62 | MDR Art. 74 (2) | MDR Art. 74 (1) | — |
| Weitere regulatorische Anforderungen | MDR Art. 62-80 + Anhang XV In Deutschland noch MPDG §§ 24–30, 62–70 | MDR Art. 62 Abs. 4b–k, m, 75–77, 80 (5) und Anhang XV + MPDG (DE) | Keine Anwendung der MDR oder des MPDG; es gelten ggf. die nationalen Vorschriften | |
| Normen/ Leitlinien | DIN EN ISO 14155 – 2021-05 Gute klinische Praxis für klinische Prüfungen von Medizinprodukten Deklaration von Helsinki, aktuelle Version von Oktober 2013, Fortaleza (Brasilien) | |||
| Leitlinien der MDCG zur klinischen Prüfung: 2023/C 163/06, MDCG 2021-28,MDCG 2021-20, MDCG 2021-8, MDCG 2021-6 MDCG 2020-10/1 MDCG 2020-10/2 | IMDRF Guidance PMCF Studies: IMDRF MDCE WG/N65FINAL:2021 | |||
| Ziel | Konformitätsbewertung | Andauernde Bewertung der Sicherheit, Leistung und des klinischen Nutzens des Produkts | ||
b) Auf nationaler Ebene
Abhängig von dem Land, in dem die klinische Prüfung durchgeführt wird, gelten länderspezifische Anforderungen.
Deutschland
Die EU-Verordnungen (MDR und IVDR) haben auch eine Erneuerung der nationalen Bestimmungen und Gesetze mit sich gebracht. Diese Anpassungen, ebenso wie spezifische nationale Festlegungen, erfolgten hauptsächlich durch das Medizinprodukte-EU-Anpassungsgesetz (MPEUAnpG). Das MPEUAnpG enthält unter Artikel 1 das Medizinprodukte-Durchführungsgesetz (MPDG), welches das bisher gültige Medizinproduktegesetz seit dem 26. Mai 2021 für Medizinprodukte, die unter die MDR fallen, ersetzt.
Notwendige Änderungen für In-vitro-Diagnostika sind in dem MPEUAnpG bereits berücksichtigt – diese treten jedoch abweichend erst zum 26. Mai 2022 in Kraft. Weitere Informationen finden Sie in unserem Blogartikel zum Medizinprodukterecht-Durchführungsgesetz (MPDG)
Der Fachartikel zum MPDG verschafft einen schnellen Überblick über das Gesetzeswerk.
Österreich
Das österreichische Bundesgesetz für die Regulation der Medizinprodukte (Medizinproduktegesetz 2021 – MPG 2021) wurde in einer neuen Fassung publiziert. Die Informationen finden Sie im Rechtsinformationssystem des Bundes (RIS), ein System, das die Gesetze in Österreich online bereitstellt.
Das konsolidierte österreichische MPG ist dem MPDG sehr ähnlich. Die wichtigen Passagen zur klinischen Prüfung sind im Abschnitt 3 §§ 13–36 hinterlegt. Weitere wichtige Gesetze finden Sie im BASG, in der Medizinproduktebetreiberverordnung und die Medizinproduktemeldeverordnung. Ähnlich wie in Deutschland gibt es auch eine österreichische Medizinproduktebetreiberverordnung.
Schweiz
Die Schweiz hat die Regelung für Medizinprodukte den EU-Regeln angepasst. Dies erfolgte im Interesse der Patientensicherheit und des EU-Marktzugangs für die Schweizer Medizinprodukte-Industrie. Die auf der Webseite des Schweizer BAG publizierte Verordnung für klinische Versuche mit Medizinprodukten (KlinV-Mep) führt alle Bestimmungen zur Forschung mit Medizinprodukten übersichtlich in einem Rechtstext auf.
Die Inkraftsetzung der angepassten Humanforschungsregelungen wurde am 26. Mai 2021 festgelegt. Auf den Webseiten von Swissmedic und swissethics findet man aktuelle Informationen zu Gesuchen über klinische Versuche mit Medizinprodukten.
Frankreich
Die nationale Richtline für Medizinprodukte in Frankreich legt die Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) fest. Die Umsetzung der MDR ist in Frankreich in der Verordnung 2022-582 ab April 2022 geregelt. Wichtig bei der Einreichung: Kennzeichnung, Konformitätserklärung und Gebrauchsanweisung von Medizinprodukten müssen gemäß dem französischen Gesetzbuch für das öffentliche Gesundheitswesen in französischer Sprache abgefasst sein.
Spanien
Die Generaldirektion für Arzneimittel und Medizinprodukte (Dirección General de Farmacia y Productos Sanitarios) ist Teil des spanischen Gesundheitsministeriums (Ministerio de Sanidad).
Der Königliche Erlass 1090/2015, der klinische Studien mit Arzneimitteln, die Ethikkommissionen für die Forschung mit Arzneimitteln und das spanische Register für klinische Studien regelt, gibt hier auch Vorgaben zum Umgang mit Medizinprodukten aus, die sich an der europäischen Rechtsprechung orientieren.
Schweden
Läkemedelsverket (LV) ist die schwedische Behörde für Medizintechnik mit Sitz in Uppsala. Sie ist die Zulassungs- und Aufsichtsbehörde für Arzneimittel in Schweden und zuständig für die Überwachung von Kosmetika, Hygieneprodukten und Medizinprodukten. Als Genehmigungs- und Aufsichtsbehörde für Betäubungsmittel regelt und überwacht sie deren Umgang und Verkehr.
Dänemark
In Dänemark ist die Danish Medicines Agency für die nationalen Richtlinien maßgebend. Die dänische Verordnung über Medizinprodukte und Produkte ohne medizinischen Zweck (BEK Nr. 957 vom 29.04.2021) ist das nationale Umsetzungsgesetz, das ab dem 26. Mai 2021 gilt. Diese Verordnung legt nationale Anforderungen fest, die Wirtschaftsakteure befolgen müssen, wenn sie in Dänemark Geräte vermarkten wollen.
Es besteht die Verpflichtung, Etiketten und Gebrauchsanweisungen in dänischer Sprache bereitzustellen, wenn das Produkt dem Endbenutzer oder Patienten zur Verfügung gestellt wird. Diese Anforderung kann jedoch in Ausnahmefällen von der dänischen Arzneimittelbehörde ausgeschlossen werden (Art. 3, Abs. 2 BEK), z. B. unter Berücksichtigung der beruflichen und sprachlichen Voraussetzungen des Benutzers für die Verwendung des Geräts mit einer Anleitung in einer Fremdsprache (meistens Englisch) und dem Verwendungszweck des Geräts.
Die Konformitätserklärung kann grundsätzlich in englischer Sprache erstellt werden. Dennoch kann die Agentur für die Marktüberwachung eine Übersetzung ins Dänische verlangen (Art. 5 BEK).
3. Typische Fehler bei der Planung von PMCF-Studien
Fehler 1: Zu viele oder die falschen Studienzentren
Die Auswahl eines passenden Studienzentrums kann sich als schwieriges Unterfangen herausstellen. Dabei kann die richtige Wahl die Datenqualität entscheidend beeinflussen. Ein kompetentes und qualitativ hochwertiges Zentrum ist die Basis einer guten Studie.
Gute Studienzentren erkennen Sie an der Ausstattung des Studienortes, der Einhaltung von vorgegebenen Zeitrahmen, hohen Kapazitäten für die Patientenrekrutierung, erfahrenen Mitarbeitern, an den Referenzen bereits getätigter Studien und schließlich auch an den aufgerufenen Kosten.
Die Zusammenarbeit mit mehreren Studienzentren kann dazu beitragen, dass Sie die benötigten Daten deutlich schneller sammeln. Machen Sie sich aber bewusst, dass jedes weitere Studienzentrum geschult, auditiert und ausgerüstet werden muss und dass Sie ihre Sponsorenpflichten für alle Zentren wahrnehmen müssen.
Fehler 2: Zu viele Studienländer
Ähnlich wie bei den Studienzentren ist auch die Wahl der Studienländer entscheidend für den zeitlichen Ablauf und die Kosten einer klinischen Prüfung. Informieren Sie sich genau über die nationalen Vorschriften und Gegebenheiten in ihren Zielländern und beachten Sie Besonderheiten bei der regionalen Einreichung.
Ziel der Studie sollte es sein, mit klinischen Daten Ihre Outcome-Parameter innerhalb Ihrer festgelegten Akzeptanzkriterien nachzuweisen. Inwieweit genetische Alterationen oder eine regionale ethnische Divergenz notwendig sind, um Ihre Studiendaten zu untermauern, können Sie aus dem Stand der Technik ermitteln.
Fehler 3: Falsches Studiendesign
Wie bei jeder qualitativ hochwertigen klinischen Studie ist es auch bei PMCF-Studien die Wahl des korrekten Studiendesigns wichtig, um Daten richtig zu erfassen. Dabei kann das Studiendesign je nach Anforderungen variiert werden (z. B. RCT-Studien oder Simulationsstudien, „Open Label“-Studien oder Cross-Over-Studien, Interventionsstudien oder Beobachtungsstudien). Welches Studiendesign für Sie das richtige ist, lässt sich nur fallspezifisch auf Ihr Produkt ausrichten. Die richtige Wahl spart Ihnen Zeit, Kosten und Nerven.
Fehler 4: Fehleinschätzung des Zeitaufwands
Viele Hersteller unterschätzen bei der Planung ihrer Studien die Zeitdauer der Patientenrekrutierung. Insbesondere bei höher klassifizierten Produkten oder wenn es um den Zugang zu den entsprechenden Patientenpopulationen und Zeitplänen geht, treten Schwierigkeiten auf. Vor allem bei seltenen Erkrankungen oder kleinen Studienpopulationen fällt das besonders ins Gewicht, da hier die Ausgangspopulationen sehr klein sein können.
Fehler 5: Unzureichende Fallzahlplanung
Um valide Ergebnisse zu erhalten, ist vor Durchführung jeder Datenerhebung eine Planung und Berechnung der Fallzahl absolut notwendig. Bei klinischen Studien und Tierversuchen ist dies sogar zwingend vorgeschrieben und wird vor Erteilung der Zulassung genau überprüft (MDR, Verordnung EU 745/2017 Anhang XV, Kapitel 1, Abschnitt 2.1; DIN EN ISO 14155:2021-05). Auch in anderen Anwendungsbereichen spricht eine fehlende Fallzahlplanung nicht für die Qualität der Studie. Versäumnisse können mit enormen Kosten oder langen Studienzeiten verbunden sein.
Die Fallzahlplanung hängt dabei von der primären Fragestellung und von der geplanten statistischen Auswertungsmethode ab. Im Vorfeld sollten eine Reihe von Annahmen bezüglich der Parameter getroffen werden (z. B. Akzeptanzkriterien, verfügbare klinische Daten, Definition von Endpunkten und statistische Stellgrößen). Jede Fallzahlplanung ist daher individuell auf die jeweilige Studie und ihre Situation zugeschnitten.
Fehler 6: Unklarheit über regulatorische Anforderungen
Bei der Fülle an Anforderungen der MDR und des MPDG an die unterschiedlichen Studientypen fällt es schwer, den Überblick zu behalten, vor allem wenn man seine ersten klinischen Prüfungen oder PMCF-Studien plant. Umfangreiche Hilfestellung bei der Umsetzung der Anforderung für die Planung und Durchführung von klinischen Studien geben das BfArM auf seiner Homepage und der Arbeitskreis der Medizinischen Ethik-Kommissionen.
Die Leitlinie des BfArM hilft, die Produkte richtig einzuordnen.
Fehler 7: Falsche Auswahl einer Clinical Research Organisation (CRO)
Clinical Research Organizations (CRO) können Sie bei allen oben genannten Problemen unterstützen. Die meisten CROs haben sich allerdings auf die Erforschung von Arzneimitteln spezialisiert.
Es ist nicht immer nötig, eine (u. U. teure) CRO in Anspruch zu nehmen. Denn nicht jedes Produkt braucht eine klinische Prüfung oder PMCF-Studie. Mehr dazu erfahren Sie in unseren Beiträgen zur MDCG 2020-6, der klinischen Bewertung oder in unserem Seminar zur klinischen Bewertung.
4. Tipps für die Planung von PMCF-Studien
Tipp 1: Bewertung der Daten standardisieren
Standardisierte Prozesse erleichtern die Datenauswertung enorm und schaffen klare Strukturen für die Technische Dokumentation und die Auditierung. Hierbei sollten die Daten über alle Geräte, medizinischen Indikationen und Zielpopulationen hinweg einheitlich organisiert werden. So lässt sich leichter erkennen, welche Daten vorhanden sind und welche noch erhoben werden müssen.
Zusätzlich trägt es dazu bei, dass alle Bereiche bei der Arbeitsteilung abgedeckt sind. Ein standardisierter Prozess erleichtert zudem die Verfolgung anderer Risikofaktoren für Hersteller, wie Reklamationstrends, Rückrufe oder Veränderungen in bestimmten Marketingregionen.
Tipp 2: Produktportfolio priorisieren
Analysieren Sie als Hersteller Ihre Medizinprodukte nach den folgenden Kriterien:
- Umsätze, Deckungsbeitrag
- Produkt-Impact im medizinischen Markt
- Ablauffristen der Zertifikate
- Ergebnisse der Gap-Analysen
- Menge der zu prüfenden Produkte in Abhängigkeit zum Lebenszyklus des Medizinprodukts
Leiten Sie daraus ab, ob die notwendigen PMCF-Aktivitäten für alle bestehenden Produktklassen wirtschaftlich sinnvoll sind.
Tipp 3: Datenmenge und Evidenzlevel bestimmen
Nachdem alle Daten erhoben sind, sollten Hersteller die Qualität und Relevanz ihrer Daten aus der Sichtweise einer Benannten Stelle hinterfragen. Bei Geräten wie Implantaten müssen Hersteller beispielsweise Daten aufnehmen, die den gesamten Lebenszyklus des Geräts abdecken.
Dies ist ein guter Zeitpunkt, um die Objektivität der gesammelten Daten zu hinterfragen und festzustellen, ob bestehende Verzerrungen möglicherweise korrigiert werden oder ob Einschränkungen der Daten im PMCF-Bericht dargelegt werden müssen.
Überprüfen Sie, ob ausreichend klinische Evidenz gesammelt wurde. Hier sind vor allem die Indikation, die Risikoklasse Ihres Produktes, die Daten zu äquivalenten und ähnlichen Produkten und die Vergleichsdaten aus dem Stand der Technik entscheidend.
Tipp 4: Betroffene Abteilungen einbeziehen
Um die langfristige Compliance zu gewährleisten, sollten Sie auch andere Abteilungen von Beginn an einbeziehen und auf dem aktuellen Stand halten. Geschäftsabteilungen, die nicht mit der Regulation der Produkte betraut sind, verstehen möglicherweise nicht vollständig, warum für etablierte Produkte weitere oder höhere Kosten anfallen.
Alle Abteilungen sollten daher einbezogen werden in Gespräche über die Produktportfolios, auftretende Risiken und alle verfügbaren Produktdatenquellen. Grundlegend sollte dabei auf potenzielle Geschäftsschäden hingewiesen werden.
Schreiben Sie eine E-Mail an die klinischen Expert:innen des Johner Instituts mit Ihrer Frage oder/und mit einem Terminvorschlag für ein kostenfreies Gespräch. Sie erhalten die notwendigen Informationen, um Ihr Projekt abschätzen und in die Planung einsteigen zu können.
5. Unterstützung bei PMCF-Studien
Die klinischen Expertinnen und Experten des Johner Instituts unterstützen Sie bei der Entscheidung, ob PMCF-Studien überhaupt notwendig sind, sowie bei der Planung und Durchführung dieser Studien.
a) Klinische Strategie für die PMCF-Studien festlegen
Falls eine Studie notwendig ist, erhalten Sie Unterstützung dabei, die klinische Strategie für Ihre PMCF-Studie und PMCF-Maßnahmen in Ihrem Clinical Evaluation Plan (CEP) festzulegen. Dazu zählen:
- Stand der Technik ermitteln und klinisch relevante Parameter ableiten
- Akzeptanzkriterien, klinischen Nutzen und die Methoden zum Nachweis definieren
- Aktuelle Datenlage bewerten und geeignete PMCF-Aktivitäten planen
- PMCF-Studien planen und vorbereiten
Somit können Sie sich sicher sein, dass Sie eine adäquate Studienplanung vorliegen haben und sich auf die richtigen Parameter und klinischen Endpunkte für Ihre PMCF-Studie konzentrieren.
b) Studiendesign erstellen und Fallzahlplanung durchführen
Welchen Studientyp Sie benötigen oder wie Sie am schnellsten zu Ihrem Ziel gelangen, nämlich der Zulassung Ihres Medizinprodukts, zeigt Ihnen die klinische Strategie. Nutzen Sie hierzu die erfahrenen Expert:innen des Johner Instituts. Sie achten darauf, dass Ihre Kosten für Studien, Fallzahlplanung und Einreichung so gering wie möglich bleiben, ohne dass Sie bei der Qualität des Nachweises des klinischen Nutzens Ihres Medizinprodukts Abstriche machen zu müssen.
Damit könnrn Sie
- die Dauer und Kosten der klinischen Prüfungen früh kennen und über die Durchführung entscheiden,
- Gewissheit erhalten, dass Ihre Benannte Stelle oder Behörde nach Studienende die Ergebnisse auch akzeptiert.
6. Fazit und Zusammenfassung
Der Gesetzgeber und die Hersteller möchten gewährleisten, dass die Medizinprodukte über den kompletten Lebenszyklus sicher, leistungsfähig und wirksam sind.
Die PMCF-Studien sind ein wichtiges Instrument, um diesen Nachweis zu führen. Das heißt aber nicht, dass PMCF-Studien immer zwingend notwendig sind. Die günstigste Studie ist die, die man nicht durchführen muss, beispielsweise, weil sich die Daten auch anders erheben lassen.
Falls eine PMCF-Studie notwendig ist, gilt es, die verschiedenen Typen zu unterscheiden und die jeweiligen regulatorischen Anforderungen zu identifizieren und zu erfüllen. Beides ist nicht ganz trivial, aber mit gezielter Hilfe gut erreichbar.
Schreiben Sie eine E-Mail an die klinischen Expert:innen des Johner Instituts mit Ihrer Frage oder/und mit einem Terminvorschlag für ein kostenfreies Gespräch. Sie erhalten die notwendigen Informationen, um Ihr Projekt abschätzen und in die Planung einsteigen zu können.


Sehr geehrter Herr Dr. Goldmann,
vielen Dank für Ihren interessanten Artikel. Inwieweit kann man das oben beschriebene auch auf IVDs gemäß IVDR übertragen?
Insbesondere würde mich interessieren, wann man keine PMPF-Sudien benötigt, bzw. diese entfallen können. Gibt es hier überhaupt geeignete Begründungen/Scenarien, wann man keine PMPF-Sudie durchzuführen braucht?
Vielen Dank und beste Grüße
Sehr geehrter Herr Resch,
Vielen Dank für Ihre spannende Anfrage! Vorab gilt, ähnlich wie es für PMCF-Aktivitäten und PMCF-Studien der Fall ist, gibt es auch weitaus mehr PMPF-Aktivitäten als PMPF-Studien. PMPF-Aktivitäten kann man selten bzw. nie vollends wegdiskutieren. Allerdings kann man je nach Etabliertheit des Produkts und der nachgewiesenen Leistung z.B. die Frequenz bestimmter Aktivitäten anpassen oder sich auf bestimmte Aktivitäten fokussieren (z.B. würde man immer eine Literaturrecherche zum Stand der Technik in der diagnostischen Praxis inkl. ähnlicher Produkten und deren Leistungsdaten durchführen, oft auch mit Ringversuche soweit Sie verfügbar sind.)
PMPF-Studien muss man tatsächlich nicht per default machen. Sie können ein sehr gutes Mittel sein, um Lücken in der Leistungsbewertung zu schließen (z.B. Fallzahl erhöhen, Subgruppen besser auswerten, RW data, etc.). Die Begründung ist – wie die gesamte Leistungsbewertungsstrategie – sehr produktspezifisch. Eine pauschale Antwort kann ich ihnen hierzu leider nicht geben, aber Ich würde Ihnen die Empfehlung nahelegen, die Details ihres Szenarios mit unseren IVD-Experten abzugleichen und in der Beratung konkret ihren Fall durchzusprechen.
Ich hoffe diese Antwort hilft ihnen weiter.
Herzliche Grüße
Dr. Johannes Goldmann
Hallo Herr Goldmann,
Vielen Dank für den umfassenden Fachartikel!
Ich habe eine Frage zu den PMCF Studien innerhalb Zweckbestimmung / ohne zusätzliche und belastende Untersuchungen („Typ 3“).
Sie ordnen diese ein als „keine Anwendung der MDR“ – so sehe ich das eigentlich auch.
Im Artikel verlinkt ist aber auch der „Leitfaden zur regulatorischen Einordnung von klinischen Prüfungen mit Medizinprodukten“. Dieser wiederum nennt diese Studien „Klinische Prüfung gemäß Art. 74 Abs. 1 S.3 MDR“ . Dies wäre also doch eine „Anwendung der MDR“ – wobei aber später im Leitfaden darauf hingewiesen wird, dass die diesbezügliche Rechtsauslegung noch diskutiert wird.
Diese Diskussion zum dritten Satz des Art 74 Abs 1 ist mir nun schon öfter begegnet.
In manchen Ländern ist die Situation ja eher klar (zB Deutschland), aber insgesamt ist die Situation natürlich unbefriedigend für diese doch häufige Studienart.
Was meinen Sie dazu? Wird es dort noch eine Klärung geben? Und interessant wäre: sollte die Auslegung „dritter Satz trifft zu“ sich durchsetzen (was ich nicht hoffe) – was würde das für diese PMCF Studien praktisch bedeuten (siehe Satz 3: Points (b) to (k) and (m) of Article 62(4), Article 75, Article 76, Article 77, Article 80(5) and the relevant provisions of Annex XV shall apply to PMCF investigations)?
Randbemerkung: Ich meine, dass es nicht die Intention gewesen sein kann, Satz 3 auf alle PMCF Studien zu beziehen. Nur als Beispiel: für die Studien ohne zusätzl Maßnahmen ist ja keine Notification der Behörde vorgesehen (sie sind explizit ausgenommen aus Satz 1 und 2 des Article 74 Abs.1). Und wenn ich nicht die Behörde vor Studienbeginn notifiziert habe (also nur bei Ethikkommissionen eingereicht habe), wie kann ich dann Art 77 befolgen (Notifizierung der Behörden zum Studienende)?
Viele Grüße und herzlichen Dank!
Sehr geehrte Frau Röthlein,
Vielen Dank, dass Sie diese interessante Fragenstellung nochmal hervorgehoben haben!
Wir haben bereits ähnlich Erfahrungen gemacht für die Einreichung von „Typ 3“ Studien, die den nationalen Vorgaben und der landespezifischen Rechtsprechung unterlegen und in unterschiedlichen Ländern verschieden interpretiert werden. Wie Sie bereits erwähnt haben ist es für Deutschland recht klar geregelt über die Registrierung der Studie und der berufsrechtlichen Beratung §15 der Ärztlichen Berufsordnung (was im Endeffekt der Anzeige und Besprechung bei der lokalen Ethikkommission entspricht). Da das Prüfungsverfahren der eingereichten Studien mittlerweile sequenziell abläuft, wird die Behörde bei solchen Studien nur noch bei der Bekanntgabe des Studienendes informiert (Zeitpunkt der Letzten Patientenvisite). Ab diesem Zeitpunkt läuft auch die angedachte Frist die Studienergebnisse innerhalb eine Jahres zu publizieren. Bei Studien nach Artikel 74, Abs. 1, Satz 3 der MDR finden Sie die Einreichungsinformationen auf Seite 22 der BfArM Anleitung https://www.bfarm.de/SharedDocs/Downloads/DE/Medizinprodukte/DMIDS-anleitung-sponsoren-mdr.pdf?__blob=publicationFile
Teils wegen sprachlicher Barrieren, teils aber auch wegen unterschiedlichen Interpretation des Art. 74 Abs. 1 S.3 MDR bestehen einige Länder für die Einreichung der Studien auf die Erfüllung der Vorgaben gemäß MDR Art. 62 Abs. 4b–k, m, 75–77, 80 (5) und Anhang XV, in Griechenland wollte sogar die Landesbehörde nachdem Ethikvotum den Antrag sequentiell prüfen, was zu einer enormen Kostensteigerung geführt hat. Deshalb haben wir in diesem Blogartikel noch einmal betont wie wichtig es ist sich mit den nationalen Gegebenheiten und Rechtsprechung vertraut zu machen, bevor man ein Studienzentrum in dem jeweiligen Land aussucht.
Der verlinkte Leitfaden, dient als erste gute Orientierungshilfe vor allem für Hersteller die vor ihrer ersten PMCF-Studie stehen. Der Leitfaden wurde als „lebendes“ Dokument konzipiert und muss an einigen Stellen noch Inhaltlich präzisiert werden um rechtliche Grauzonen deutlicher zu definieren. Ich teile aber vollends ihre Meinung, dass die aktuelle Situation eher unzufriedenstellend gelöst wurde. Die länderspezifischen Probleme in der Einreichung könnten mit einer gesamteinheitlichen europäischen Lösung deutlich reduziert werden. Für den Arzneimittelbereich konnte diese Transition wie klinische Prüfungen in der Europäische Union (EU) durchgeführt werden, mit der Einführung der Verordnung über klinische Prüfungen (Verordnung (EU) Nr. 536/2014) die am 31.01.2022 in Kraft getreten ist deutlich vereinfacht werden. Mit dieser Verordnung werden die Einreichungs-, Bewertungs- und Überwachungsverfahren für klinische Prüfungen in der EU über das Informationssystem für klinische Prüfungen (Clinical Trials Information System – CTIS) vereinheitlicht. Ein solcher Schritt wäre auch für den Bereich der klinischen Prüfungen von Medizinprodukten wünschenswert. Ob die Einführung der Module für klinische Prüfungen und die klinische Nachbeobachtung der EUDAMED das Problem lösen können, bleibt weiterhin abzuwarten.
Ich danke ihnen aber für diesen wertvollen Input und hoffe das hier in naher Zukunft eine
deutlichere Klärung möglich ist.
Herzliche Grüße und ein schönes Wochenende
Dr. Johannes Goldmann
Hallo Herr Goldmann,
ich bin gerade auf den Satz: „When the manufacturer plans to have the first report. The timelines shall be defined quarterly or at least yearly.“ im MDCG 2020-7 gestoßen. Dieser doch etwas unglücklich formuliert… Auf was beziehen sich die genannten Zeiträume? Können Sie mir in diesem Zusammenhang Auskunft geben?
Vorab vielen Dank!
Sehr geehrte Frau Berg,
Vielen Dank für ihre Frage !
Ich gebe ihnen Recht, dass die Formulierung hier etwas Raum für Interpretationen bietet. Von unserem Verständnis definieren Sie als Hersteller in ihrem PMCF-Plan ihre Aktivitäten, die notwendig sind, um eine adäquate Aktualisierung ihrer klinischen Bewertung zu ermöglichen. Je nach Profil und Risiko des Medizinproduktes legen Sie hierfür einem zeitlichen Turnus für ihre PMCF-Maßnahmen(z.B. systematische Kundenbefragungen, PMCF-Studien, PMCF-Surveys etc.) fest. Bewegen Sie sich zum Beispiel in einem sehr volatilen Markt mit starken Entwicklungssprüngen wäre es sinnvoll die Aktualisierung der Literatur zum Stand der Technik zu ihrem Produkt in einem kurzen Zeit Intervall zu beobachten und in einem PMCF-Bericht zu bewerten. Ein Minimum der Aktualisierung des PMCF-Berichts wird hier aber auf jährlich gesetzt, da Sie mit dem PMCF-Maßnahmen ja die Anwendung ihres Produktes unter Real-Life Bedingungen im Markt überwachen möchten. Bringt ihnen diese Ausführung mehr Klarheit ? Ansonsten kommen Sie gerne auf unsere Experten im Clinical Affairs Bereich zu, wir helfen ihnen auch die Anforderungen direkt auf ihre Situation und ihr Produkt anzupassen.
Herzliche Grüße
Dr. Johannes Goldmann