
Die MDR verpflichtet die Hersteller von Medizinprodukten zur Post-Market Surveillance (PMS) und damit zum Sammeln von PMS-Daten. Dieser Artikel soll zur Klärung beitragen, welche regulatorischen Anforderungen Hersteller beim Sammeln dieser PMS-Daten beachten müssen. Insbesondere soll er aufzeigen, ob Hersteller eine Ethik-Kommission einbeziehen müssen, wenn die Daten bei der Routine-Anwendung der Produkte anfallen.
1. PMS-Daten: Die Fragestellung
Auslöser für diesen Artikel war eine Diskussion zwischen einem Hersteller medizinischer Software und einer Benannten Stelle. Beide waren sich uneins über die Notwendigkeit, beim Sammeln von PMS-Daten die Deklaration von Helsinki zu beachten und ein Ethikvotum einzuholen. Es herrschte auch Dissens darüber, ob dieses Sammeln von Daten bereits eine klinische Prüfung sei.
Im konkreten Fall nutzte der Hersteller Daten für die Post-Market Surveillance, die bei der Routinenutzung des Produkts anfielen. Konkret: Der Hersteller griff weder in die Behandlung ein, noch erfasste er zusätzliche Daten. Vielmehr nutzte er die Daten, die das Medizinprodukt bei der zweckbestimmungsgemäßen Nutzung in seiner Datenbank speicherte.
Die zu klärende Hauptfrage lautet: „Wann benötigt ein Hersteller beim Sammeln von PMS-Daten ein Ethikvotum, und wann zählt dieses Sammeln als klinische Prüfung?“
Um diese Fragen zu beantworten, muss sie in weitere Teilfragen zerlegt werden. Weiter unten finden Sie diese Teilfragen und die zugehörigen Antworten.
2. Relevanz von PMS-Daten
Mit dem Übergang auf die MDR rückt das Sammeln von PMS-Daten mehr ins Bewusstsein der Hersteller. Benannte Stellen fordern die PMS-Pläne ein und wollen verstehen, wie die Daten gesammelt, ausgewertet sowie bewertet werden und wie sie Eingang in u. a. die klinische Bewertung finden.
Falls Hersteller die regulatorischen Hürden für das Sammeln von PMS-Daten (irrtümlich) als zu hoch einschätzen, kann das dazu führen, dass sie Routinedaten nicht verwenden. Damit konterkarieren sie die Zielsetzung der MDR, durch Post-Market-Daten belastbare Aussagen zur Sicherheit und Leistungsfähigkeit der Medizinprodukte zu erlangen.
Wenn Klarheit über die regulatorischen Anforderungen herrscht, können Hersteller unnötige regulatorischen Risiken (bei der „Zulassung“) vermeiden. Zudem blieben Herstellern und Benannten Stellen unnötige und zeitintensive Diskussionen erspart – und den Ethik-Kommissionen unnötige Anfragen.
3. Antworten auf die Fragen zum Sammeln der PMS-Daten
Disclaimer & Acknowledgement
An diesem Artikel haben Juristen einer deutschen Behörde und der EU mitgewirkt, die aber nicht genannt werden wollen. Die Darstellungen in diesem Artikel sind „in progress“, da auch die Juristen untereinander noch nicht in allen Punkten Übereinstimmung erzielt haben.
Annahmen
Die Antworten gehen von den folgenden Voraussetzungen aus:
- Der Hersteller sammelt Daten (bzw. lässt diese sammeln, z. B. durch den Betreiber oder den Anwender). Diese Daten nutzt er für die Post-Market Surveillance.
- Die Daten fallen „automatisch“ bei der zweckbestimmungsgemäßen und routinemäßigen Verwendung des Produkts an. Und das auch unabhängig davon, ob sie für die Post-Market Surveillance benötigt werden. Das heißt, dass für die PMS keine zusätzlichen Daten erhoben werden.
- Es finden keine zusätzlichen oder gar die Patienten belastenden Verfahren statt.
- Die gesammelten Daten lassen sich den Personen (Patienten) zuordnen.
Beachten Sie, dass alle Aussagen in diesem Artikel von diesen Voraussetzungen ausgehen.
Zusammenfassung
Das Schaubild gibt in Kurzform einen Überblick über Fragen und Antworten:
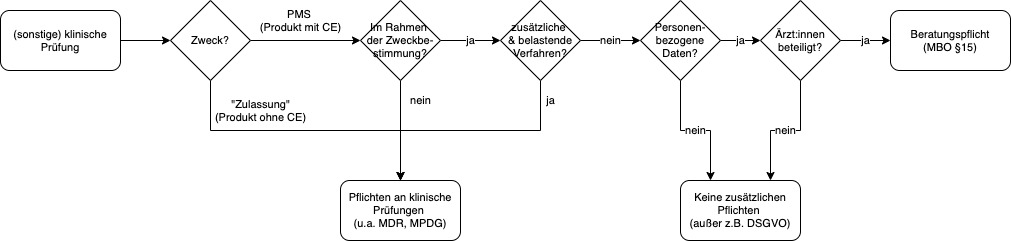
Beim Sammeln von PMS-Daten unter den o. g. Voraussetzungen greifen die Anforderungen der MDR an klinische Prüfungen nur sehr eingeschränkt. Es besteht keine Genehmigungspflicht durch Ethik-Kommissionen.
Der Datenschutz muss immer gewährleistet sein.
Das BfArM schreibt sogar (Seite 25): „Weder in der MDR noch im MPDG werden Anforderungen an PMCF-Studien definiert. (Lesart des letzten Satzes in Artikel 74 Absatz 1 der MDR: nicht allgemeingültig)“
Frage 1: Handelt es sich bei den Daten um klinische Daten?
Antwort: Ja
Begründung: Es handelt sich per definitionem (MDR Artikel 2 (48), vierter Spiegelstrich) um klinische Daten.
Frage 2: Liegt eine klinische Prüfung vor?
Antwort: Umstritten
Begründung: Einige Juristen vertreten die Meinung, dass gemäß MDR Art. 2 (45) eine klinische Prüfung vorliegt, weil bei der Anwendung des Produkts im Rahmen seiner Zweckbestimmung menschliche Prüfungsteilnehmer einbezogen sind und weil das Ziel der PMS darin besteht, die Sicherheit oder Leistung des Produkts (nach der Inverkehrbringung) zu bewerten.
Es gibt jedoch auch Argumente gegen diese Einschätzung. So ist es zum Beispiel umstritten, ob die Patienten als Prüfungsteilnehmer anzusehen sind.
Selbst wenn das Sammeln von PMS-Daten definitionsgemäß als klinische Prüfung zu verstehen wäre, folgt daraus noch nicht, dass die Anforderungen der MDR an die klinischen Prüfungen zu beachten sind (siehe auch Frage 4).
Das Ziel der MDR besteht darin, Patienten vor unsicheren Medizinprodukten und unethischen klinischen Prüfungen zu schützen. Beim Sammeln von PMS-Daten bestehen solche Gefahren nicht.
Frage 3: Liegt eine sonstige klinische Prüfung vor?
Antwort: Nein
Begründung: Eine sonstige klinische Prüfung im Sinne des MPDG liegt nicht vor, weil das Sammeln von PMS-Daten ein systematischer und geplanter Prozess zur Produktbeobachtung ist und gemäß MDR erfolgen muss. Genau das schließt das MPDG in seiner Definition „sonstiger klinischer Prüfungen“ aus (MPDG § 3 Nr. 4 Buchstabe a)).
Frage 4: Müssen die Anforderungen der MDR Art. 62 ff. eingehalten werden?
Antwort: Ja, allerdings nur einige Abschnitte
Begründung: Die Anforderungen von Art. 62 der MDR greifen nur dann vollständig, wenn die klinische Prüfung als „Teil der klinischen Bewertung für Konformitätsbewertungszwecke“ durchgeführt wird.
Artikel 74 befasst sich mit klinischen Prüfungen von Medizinprodukten, die bereits ein CE-Kennzeichen tragen. Er verpflichtet die Sponsoren zur Einhaltung einiger Abschnitte von Art. 62, selbst wenn die Patient:innen keinen zusätzlichen und invasiven oder belastenden Verfahren unterzogen werden und das Produkt im Rahmen seiner Zweckbestimmung eingesetzt wird.
Diese Anforderungen (u. a. Artikel 62 Abs. 4 Buchstaben b bis k und m) stellen allgemeine ethische und regulatorische Grundsätze dar. Sie verpflichten den Hersteller aber nicht zu einem Antrag auf Genehmigung einer klinischen Prüfung.
Frage 5: Welche weiteren regulatorischen Anforderungen sind zu beachten?
Antwort: Neben den datenschutzrechtlichen Vorgaben (DSGVO bzw. BDSG) sind in Deutschland die Berufsordnungen der jeweiligen Ärztekammern zu beachten.
Zudem müssen die Hersteller die Anforderungen erfüllen an
- die Überwachung nach dem Inverkehrbringen (Post-Market Surveillance PMS) gemäß Art. 83 sowie Anhang III und
- die klinische Nachbeobachtung nach dem Inverkehrbringen (Post-Market Clinical Follow-up PMCF) gemäß Art. 61 (11) sowie Anhang XIV Teil B.
Daraus ergaben sich die in diesem Dokument beleuchteten Fragestellungen.
Begründung: Die PMS-Tätigkeiten, an denen sich Ärztinnen und Ärzte beteiligen, unterliegen dem § 15 der Berufsordnungen ihrer jeweiligen Ärztekammer. Diese sind anwendbar, weil personenbezogene Daten gesammelt werden. Dieser Paragraf verpflichtet die Ärztinnen und Ärzte zu einer Beratung(!) durch eine Ethik-Kommission.
Ob die Daten ohnehin bereits gesammelt wurden, ist unerheblich. Entscheidend ist, dass die Daten, die im Rahmen der normalen Nutzung des Produkts anfielen, jetzt für Forschungszwecke verwendet werden sollen.
Wenn keine Ärztinnen und Ärzte beteiligt sind, weil z. B. der Hersteller die Daten direkt von den Patient:innen bezieht (was bei vielen „Wearables“ der Fall ist), greifen die Berufsordnungen der Ärztekammern und die darüber referenzierte Deklaration von Helsinki nicht. Beachten Sie jedoch den Hinweis bei Frage 6.
Zudem unterliegen auch die beim Hersteller mit der Studiendurchführung beschäftigten Ärztinnen und Ärzte den Pflichten der Berufsordnungen, sofern sie Mitglieder einer deutschen Ärztekammer sind.
Die einschlägigen datenschutzrechtlichen Vorschriften (insbesondere DSGVO und BDSG) gelten unabhängig von und zusätzlich zu den berufsrechtlichen Vorschriften.
Demnach müssen die Hersteller die Grundsätze der Datenverarbeitung beachten wie die Zweckgebundenheit, die Datenminimierung und die Minimierung der Verarbeitungsdauer (s. DSGVO Art. 5 Abs. c). Auch müssen sie den Pflichten als „Verantwortliche“ nachkommen und beispielsweise technische und organisatorische Maßnahmen zum Schutz der Daten gewährleisten.
Frage 6: Muss eine Ethik-Kommission einbezogen werden?
Antwort: In der Regel ja, aber nicht für eine Genehmigung bzw. ein Votum
Begründung: Wie in der Antwort auf die letzte Frage ausgeführt, müssen die Ärztinnen und Ärzte in den genannten Fällen eine Beratung durch die Ethik-Kommission nach der Berufsordnung der jeweiligen Ärztekammer einholen. Sie sind aber nicht zu einer Genehmigung durch eine Ethik-Kommission gemäß MDR verpflichtet.
Diese Pflicht betrifft auch die beim Hersteller mit der Studiendurchführung beschäftigten Ärztinnen und Ärzte, sofern sie Mitglieder einer deutschen Ärztekammer sind.
Auf den Seiten des BfArMs waren die Einschätzungen des Arbeitskreises medizinischer Ethik-Kommissionen publiziert. Diese sind leider nicht mehr abrufbar. Die folgende Grafik lehnt sich an die damals veröffentlichen Inhalte an.

Diese Darstellungen kommen zum gleichen Ergebnis: Beim Sammeln von PMS-Daten gilt unter den o. g. Voraussetzungen:
- Die Anforderungen an klinische Prüfungen, insbesondere Art. 62 der MDR, greifen nur eingeschränkt.
- Es handelt sich nicht um eine „sonstige klinische Prüfung“.
- Es besteht keine Genehmigungspflicht bei der Bundesoberbehörde.
- Die ärztlichen Berufsordnungen müssen beachtet werden, wenn Ärztinnen oder Ärzte beteiligt sind. Es besteht jedoch keine Genehmigungspflicht durch eine Ethik-Kommission.
Beachten Sie, dass die Deklaration von Helsinki auch andere Personen als Ärztinnen und Ärzte adressiert: Der WMA regt andere an der medizinischen Forschung am Menschen Beteiligte an, diese Grundsätze zu übernehmen.
Frage 7: Ist die ISO 14155 anwendbar?
Antwort: Ja, aber nicht alle Teile
Begründung: Die ISO 14155 ist für klinische Prüfungen anwendbar. Sie beinhaltet „klinische Beobachtungsstudien“ explizit im Anwendungsbereich. An diese Beobachtungsstudien stellt die ISO 14155 niedrigere Anforderungen.
Welche Ziele der Gesetzgeber verfolgt, lässt sich auch aus der IVDR ableiten. Sie erlaubt aus „diagnostischen Routinetests gewonnene Erfahrungen“ zum Nachweis der klinischen Leistung eines IVDs zu nutzen. Auch für diese Daten fordert die EU-Verordnung, die ethischen und regulatorischen Grundlagen zu erfüllen. Allerdings ist für IVD nicht die ISO 14155, sondern die ISO 20916 anwendbar. Letztere beschreibt die gute Studienpraxis für „klinische Leistungsuntersuchungen an menschlichem Untersuchungsmaterial“.
Frage 8: Schafft die MDR die gesetzliche Voraussetzung für eine DSGVO-konforme Datenverarbeitung beim Hersteller?
Antwort: Nein
Begründung: Art. 6 Abs. c) der DSGVO erlaubt zwar die Datenverarbeitung, falls „die Verarbeitung zur Erfüllung einer rechtlichen Verpflichtung erforderlich ist, der der Verantwortliche unterliegt“. Eine den Anforderungen des Art. 6 Abs. 3 DSGVO entsprechende mitgliedstaatliche oder unionsrechtliche gesetzliche Regelung, die eine Befugnis zur Datenverarbeitung für den Zweck der PMS bzw. PMCF schafft, existiert aber nicht.
Diese folgt insbesondere nicht aus der Definition des Begriffs der Überwachung nach dem Inverkehrbringen (MDR Art. 2, Nr. 60), dem Begriff der klinischen Daten (MDR Art. 2, Nr. 48) oder Art. 61, Art. 83, Anhang III und XIV Teil B MDR, den Regelungen des MPDG und auch nicht aus dem Begriff der Datenverarbeitung (DSGVO, Art. 4 Nr. 2).
Zudem müssen die Hersteller beachten, dass die klinischen Daten bzw. Post-Market-Daten Gesundheitsdaten im Sinne des Art. 4 Nr. 15 DSGVO sind. Deren Verarbeitung ist nur unter den in der DSGVO Art. 9 Abs. (2) genannten Voraussetzungen möglich. Hier ist besonders der Buchstabe i) relevant:
„Die Verarbeitung ist aus Gründen des öffentlichen Interesses im Bereich der öffentlichen Gesundheit, wie […] zur Gewährleistung hoher Qualitäts- und Sicherheitsstandards bei der Gesundheitsversorgung und bei Arzneimitteln und Medizinprodukten, auf der Grundlage des Unionsrechts oder des Rechts eines Mitgliedstaats, das angemessene und spezifische Maßnahmen zur Wahrung der Rechte und Freiheiten der betroffenen Person, insbesondere des Berufsgeheimnisses, vorsieht, erforderlich […]“
Eine entsprechende mitgliedstaatliche oder unionsrechtliche gesetzliche Regelung, die eine Befugnis zur Datenverarbeitung nach Art. 9 Abs. 2 lit. i) DSGVO für den Zweck der PMCF schafft, existiert aber nicht.
4. Konkrete Stellungnahmen
An dieser Stelle sammeln wir Stellungnahmen von Ethik-Kommissionen, Behörden, Benannten Stellen und Herstellern.
a) Einschätzung einer Landes-Ethik-Kommission
Die folgende Einschätzung stammt aus dem Dezember 2022:
Wenn das zu prüfende Medizinprodukt eine CE-Kennzeichnung trägt und im Rahmen der Studie keine zusätzlich belastenden oder invasiven Maßnahmen erfolgen und auch nicht einem Prüfplan gefolgt wird, handelt es sich um eine sonstige Studie nach § 15 der BO für Ärzte, für die nur unter folgenden Voraussetzungen eine berufsrechtlichen Beratung durch die Ethik-Kommission bei der Landesärztekammer Baden-Württemberg notwendig/möglich ist:
- Es handelt sich um ein Forschungsvorhaben,
- an dem sich ein Arzt beteiligt und
- bei dem in die psychische oder körperliche Integrität eines Menschen eingegriffen oder Körpermaterialien oder Daten verwendet werden, die sich einem bestimmten Menschen zuordnen lassen.
Sofern die Daten anonym (d. h. ohne die Möglichkeit, sie einer bestimmten Person zuordnen zu können) erhoben werden und ansonsten nicht in die psychische oder körperliche Integrität eines Menschen eingegriffen wird (insb. durch Untersuchungen), unterliegt das Forschungsvorhaben nicht der berufsrechtlichen Beratung durch die Ethik-Kommission.
Anmerkung: Dieser Beitrag geht von der Annahme aus, dass personenbezogene Daten erfasst werden.
5. Fazit & Zusammenfassung
In den meisten Fällen ist beim Sammeln von patientenbezogenen PMS-Daten keine zustimmende Bewertung durch eine Ethik-Kommission notwendig, jedoch eine Beratung durch die Ethik-Kommissionen der Ärztekammern oder Universitäten.
Diese Beratung ist zwar nicht rechtlich bindend. Allerdings setzen vielen (höherrangige) Journals für die Publikation eine Zustimmung durch eine Ethik-Kommission voraus. Würde man also deren Rat nicht folgen, wäre diese Voraussetzung nicht erfüllt.
Die Beratung erfolgen überwiegend innerhalb von 3 bis 4 Wochen.
Die Hürde durch diese Beratung sollte somit für die Hersteller kein Anlass sein, um der Verpflichtung zum Sammeln und Auswerten von Post-Market-Daten (insbesondere PMCF-Daten) nicht nachzukommen. Vielmehr sollten Hersteller die Beratung als Hilfestellung verstehen, um die Anforderungen an den Datenschutz einzuhalten.
Der Wunsch an die Entscheidenden bei Behörden, Benannte Stellen und Ethik-Kommissionen lautet:
- Behalten Sie den ursprünglichen Sinn der Regularien im Fokus.
- Überinterpretieren Sie regulatorische Anforderungen nicht. Folgen Sie gerne der Einschätzung des BfArMs:„Weder in der MDR noch im MPDG werden Anforderungen an PMCF-Studien definiert. (Lesart des letzten Satzes in Artikel 74 Absatz 1 der MDR: nicht allgemeingültig“)
- Helfen Sie den Hersteller dabei, PMS-Daten ohne unnötige Hürden zu sammeln. Denn wenn diese aus Angst vor Ihnen darauf verzichten, erreichen Sie das Gegenteil dessen, was das Ziel der Regulierung war: Mehr Patientensicherheit.
Das Johner Institut unterstützt Medizinproduktehersteller
- beim Erstellen und Prüfen von PMS- und PMCF-Plänen,
- beim automatisierten Sammeln von Post-Market-Daten,
- bei klinischen Bewertungen und Leistungsbewertungen.
Änderungshistorie
- 2023-01-24: Stellungnahme einer Ethikkommission eingefügt



Liebes Team Johner Institut,
vielen Dank für den Artikel und den Fokus auf dieses drängende und sehr relevante Praxisthema. Einen Hinweis würde ich gerne noch ergänzen: Das BfArM hat eine Interpretation zur Anwendbarkeit der MDR Anforderungen an klinische Prüfungen, wonach für Studien mit zugelassenen Produkten, gemäß Zweckbestimmung und ohne zusätzliche belastende oder invasive Verfahren für den Patienten keine MDR Anforderungen anzuwenden sind (BfArM Hinweis: Lesart des letzten Satzes in Artikel 74 Absatz 1 der MDR: nicht allgemeingültig).
Quelle: https://www.bfarm.de/SharedDocs/Downloads/DE/Service/Termine-und-Veranstaltungen/dialogveranstaltungen/dialog_2021/210505/3_schriever.pdf?__blob=publicationFile (siehe Folie 25 und 26).
Unsere Benannte Stelle (TÜV Süd) toleriert diese Interpretation bzw. unsere Anwendung der Interpretation bisher.
Sehr geehrter Herr Schuster,
das ist ein so wichtiger Hinweis. Vielen Dank!
Ich ergänze den Artikel sofort um diese Veröffentlichung.
Nochmals besten Dank!
Herzliche Grüße, Christian Johner
Liebes Johner Institut,
vielen Dank für das Aufgreifen einer sehr aktuellen Thematik, welches uns auch gerade beschäftigt.
Wir haben in unserer Recherche viel mit dem MDCG 2021-6 Entscheidungsbaum auf Seite 17 versucht zu arbeiten, wobei dieser besondere Fall entweder unter Artikel 82 MDR fällt oder durch den Entscheidungsbaum nicht abdeckt wurde (vgl. MDCG 2021-6, S.17).
Das Central Comittee on Research Involving Human Subject hat sich – wie auch das BfArM – gegen die Anwendbarkeit der MDR bei oben angesprochenen Studien ausgesprochen. Sie beziehen sich explizit auf MDCG 2021-6:
„PMCF studies which are non-interventional, for instance clinical data are obtained by file research and no additional invasive or burdensome procedures compared to standard of care are applied, fall outside the scope of chapter VI of the MDR and outside the scope of the Medical Research Involving Human Subjects Act (WMO) and are considered non-WMO studies.“
https://english.ccmo.nl/publications/publications/2021/05/17/explanatory-note-to-flowchart-to-determine-which-mdr-article-is-applicable-to-the-clinical-investigation
Sie hatten im Fall 2 „Prüfungsteilnehmer“ angesprochen. Haben Sie hier eine offizielle Definition? Wenn wir unseren DICOM-Viewer auf einem Public-Dicom-Dataset von Ärzten evaluieren lassen, sind keine Patienten beteiligt, aber vll. könnten Ärzte als Prüfungsteilnehmer verstanden werden. Haben Sie hier Erfahrungen gemacht?
Lieber Herr Kasan,
verzeihen Sie die langsame Antwort. Über uns ist eine Erkältungswelle hereingebrochen, weshalb viele Kolleginnen und Kollegen ausfielen.
Sie fragen, ob Ärzte als Prüfungsteilnehmer zu verstehen sind. Die kurze Antwort lautet „nein“.
Die längere Antwort lautet: All die in diesem Artikel diskutierten Regularien dienen dem Schutz von Patienten. Sei es der Schutz vor unsicheren Medizinprodukten, sei es der Schutz vor unnötigen Belastungen, sei es der Schutz vor unrechtmäßiger Nutzung der Daten, sprich Datenschutz.
Die Forderungen beziehen sich nicht auf die durchführenden dieser Prüfungen, weder auf die Sponsoren, noch auf an den Prüfungen beteiligten Ärztinnen und Ärzten.
Viele Grüße
Christian Johner
Liebes Johner Team.
Recht herzlichen Dank für den wieder mal hochinteressanten und perfekt recherchierten Artikel. Als Facharzt für Anästhesie / Notfallmedizin und Sachverständiger für MP werde ich regelmäßig von Herstellern angefragt, ob wir mit unserem Team PMCF Studien durchführen können.
Aus diesem Grund haben wir uns aktuell intensiv mit diesem Thema beschäftigt und werden in absehbarer Zeit die ersten Anfragen nach §15 MBO an die zuständigen Ärztekammern stellen. Sobald wir dahingehend weitere Erfahrungen und Informationen erhalten, werden wir Ihnen diese gerne zur Verfügung stellen.
Auf jeden Fall ist es für ärztliche Kolleginnen und Kollegen ratsam, einen Kurs als „Prüfarzt“ zu absolvieren, um die formalen Voraussetzungen für die Betreuung und Durchführung von klinischen Prüfungen jedweder Art zu erlangen. Entsprechende Kurse (online) bietet z.B. das Zentrum für klinische Studien der Universität Freiburg an. Infos unter: https://www.uniklinik-freiburg.de/zks/fortbildungen.html
Liebes Team des Johner Instituts, liebe Kommentierenden,
auch von mir viele Dank für diese wichtige Aufbereitung des Themas. In der Tat hat uns die MDR hier einen enormen Graubereich beschert. Wir erleben aktuell beim Umgang mit diesem Thema auch im Ausland unterschiedlichste Interpretationen und Herangehensweisen.
Noch eine Nachfrage zur Einordnung des Kommentars von Herrn Schuster bzw. der BfArM-Interpretation: Wenn das BfArM von fehlender „Allgemeingültigkeit“ spricht, meint es dies vor dem Hintergrund, dass MDR74(1) in Satz 1 eben nur solche PMCF-Studien mit zusätzlichen, invasiven und belastenden Verfahren berücksichtigt, nicht-interventionelle Studien aber außen vor lässt und somit Satz 3 sich auch nur auf interventionelle PMCF-Studien beziehen kann, richtig?
Müsste es dann in der Begründung zu Frage 4 oben statt „Artikel 74 … verpflichtet die Sponsoren zur Einhaltung einiger Abschnitte von Art. 62, selbst wenn die Patient:innen keinen zusätzlichen und invasiven oder belastenden Verfahren unterzogen werden … .“ nicht heißen: „Artikel 74 … verpflichtet die Sponsoren zur Einhaltung einiger Abschnitte von Art. 62, wenn die Patient:innen zusätzlichen und invasiven oder belastenden Verfahren unterzogen werden … .“ ?
Lieben Dank für Ihre Mühen und beste Grüße!
Lieber Herr Gesing,
bitte verzeihen Sie unsere extrem langsame Reaktion auf ihre wichtige Frage.
Wenn Patienten „zusätzlichen und invasiven oder belastenden Verfahren unterzogen werden“ greift der Artikel 62 sowieso. Das war allerdings nicht der Scope des Artikels. Dieser beleuchtet genau den Fall, dass keine „invasiven oder belastenden Verfahren“ gibt.
Das (zumindest für viele) Unerwartete ist, dass der Artikel 62 selbst in Fällen nicht ganz obsolet wird, in denen die Patienten überhaupt nicht belastet werden. Es verbleiben allerdings keine gravierenden Anforderungen.
Beste Grüße, Christian Johner
Sehr geehrtes Johner Team,
vielen Dnk für diese klärenden Artikel. In Deutschland scheint zumindest geklärt zu sein, dass bei PMCF-Studien ohne zusätzliche invasive oder belastende Verfahren oder ganz ohne zusätzliche Verfahren keine zustimmende Bewertung der Ethikkommission und/oder der BOB benötigt wird. Wie sieht dies in anderen Ländern der Union aus? Wird dies in jedem Land separat bzw. rein national reguliert? Existiert hierzu vielleicht sogar eine Übersicht? Was ist, wenn der Medizinproduktehersteller nicht in der Union ansässig ist, benötigt er dann wie bei klinischen Prüfungen einen rechtlichen Vertreter mit Sitz in der Union?
Lieben Dank und viele Grüße!
Liebe Frau Gasch,
danke für Ihre wichtige Frage, wie die Situation in anderen Ländern aussieht.
Ich verfüge leider noch nicht über diesen Überblick. Es gibt jedoch einige Anhaltspunkte, dass es vergleichbar geregelt ist:
Allerdings erwarte ich, dass sich die Überwachungspraxis unterscheidet – was sie sogar innerhalb der deutschen Bundesländer tut.
Beste Grüße, Christian Johner
Liebe Frau Gasch,
wir haben diesbezüglich relativ intensiv mit Blick auf eine nicht-interventionelle Medizinproduktestudie mit einem CE zertifizierten MP der Klasse III recherchiert – ich kann bestätigen, dass man in diesem Fall auf eine Sammelsurium nationaler Regelungen trifft. Die regulatorischen Anforderungen erstrecken sich von der Einreichung analog einer MDR 74 Studie bis hin zu zum völligen Wegfallen zur Verpflichtung des Hinzuziehens irgendeiner regulatorischen Institution. Es sind jeweils aufwendige Recherchen der nationalen Anforderungen notwendig.
Vielleicht hilft das ja schon etwas.
Viele Grüße
Stefan Gesing
Guten Morgen.
Recht herzlichen Dank für die verschiedenen Betrachtungsweisen aus den unterschiedlichen Blickwinkeln. Vielleicht sehe ich das etwas zu pragmatisch aber wir führen unsere PMCF Studien, für die Generierung von klinischen Daten, für die von uns PMS betreuten Produkte, an denjenigen Standorten durch, an den der wenigste regulatorische Aufwand entsteht. Meines Wissens nach gibt es keine Vorgabe, wo die benötigten klinischen Daten generiert werden müssen. Dieses Procedere würde ich gerne zur Diskussion stellen.
Danke vorab
Grüße
Thomas Castner
Sehr geehrter Herr Dr. Castner,
Danke für Ihre Gedanken!
Der Ansatz, die Freiheitsgrade in unterschiedlichen Rechtsbereichen zu nutzen, ist sicher hilfreich. Dennoch einige Gedanken dazu:
Beste Grüße, Christian Johner
Sehr geehrtes Johner-Team,
Ganz herzlichen Dank für die übersichtliche Darstellung dieses komplexen Themas!
Ich möchte eine Frage stellen, die an Ihre Ausführungen anknüpft: Erfordert eine Datensammlung nach Ihren Vorraussetzungen Aufklärungen und Einwilligung der Patienten, deren Daten anonym gesammelt werden?
1. Eine Aufklärung über einen geplanten Eingriff. Diese Aufklärung ist grundsätzlich Voraussetzung für die Durchführung des Eingriffs — auch ohne Datensammlung. Ist aber eine besondere Einwilligung nach Aufklärung erforderlich, die sich insbesondere auf das zu beobachtende Medizinprodukt bezieht?
2. Eine Aufklärung über die Weitergabe der anonymisierten Daten an Dritte (die Firma, die das Medizinprodukt vertreibt).
Ganz herzlichen Dank für Ihre Einschätzung!
Beste Grüße,
Christoph Schmitz
Lieber Kollege Schmitz,
danke für Ihr Nachfragen!
Der „Scope“ des Artikel beschränkt sich ausschließlich auf nicht-interventionelle Maßnahmen, sprich nicht(!) auf Eingriffe.
Ich stimme Ihnen absolut zu, dass bei Eingriffen Aufklärungen notwendig sind.
Eine Weitergabe anonymisierter Daten bedarf hingegen keiner Aufklärung oder gar Genehmigung. Für diese greifen die Datenschutzvorschriften nicht mehr, auch nicht die Deklaration von Helsinki und die daraus abgeleiteten MBOs. Vorausgesetzt der erwähnte Scope…
Beste Grüße, Christian Johner
Liebes Johner Institut,
vielen Dank für diesen klärenden Beitrag.
Verstehe ich das richtig? Eine Anwenderbefragung, deren Zielstellung im Einklang mit PMCF-Zielen (Anhang XIV, Teil B, Punkt 6.1 MDR) steht, ohne zusätzliche invasiven/belastenden Verfahren und ohne Beteiligung eines Arztes bedarf keine Genehmigung oder Anzeige bei der Bundesoberbehörde (Beratung durch die EK)?
Ist überhaupt eine Befragung bestehender Anwender/Endkunden als klinische Prüfung nach dem Inverkehrbringen zu betrachten? Wie verhält es sich, wenn Produkttester mit dem CE-gekennzeichneten Produkt einbezogen werden?
Was ist wenn das Medizinprodukt bei bestimmungsgemäßer Verwendung in den Körper gelangt? Meiner Meinung nach geht der invasiven Charakter des Medizinproduktes in diesem Fall nicht über die normale Anwendung hinaus, so dass Art. 74 nicht zutreffend ist. Wie sehen Sie das?
Beste Grüße
Sehr geehrte Frau Kouam,
besten Dank für Ihre Frage!
Eine Anwenderbefragung zählt nicht als klinische Prüfung und bedarf daher auch keiner Zustimmung durch eine Ethikkommission. Daran ändert sich auch nichts, wenn ein Produkttester einbezogen ist.
Mir ist noch nicht ganz klar, welchen Bezug Ihre zweite Frage zur ersten hat. Geht es weiter um die Anwenderbefragung? Welche Daten werden bei der bestimmungsgemäßen (invasiven) Verwendung erhoben? Ich fehlt mir noch etwas das Verständnis, um zielführend antworten zu können.
Mit den besten Grüßen, Christian Johner
Sehr geehrter Herr Johner,
vielen lieben Dank für Ihre Rückmeldung.
Es geht bei der zweiten Frage weiterhin um die Befragung, aber dieses Mal nicht von bestehenden Kunden. Produkttester werden rekrutiert, um Fragen zu beantworten, nachdem sie die bereitgestellten Medizinprodukte benutzt haben.
Bei der bestimmungsgemäßen (invasiven) Verwendung werden Fragen zu den ausgelobten Eigenschaften der besagten Produkte gestellt. Ziel ist es somit mehr klinische Daten zu generieren, die dann in den CER-Bericht einfließen können, um u.a. die Leistung des Produkts während seiner erwarteten Lebensdauer zu bestätigen, oder zuvor unbekannte Nebenwirkungen zu ermitteln und die ermittelten Nebenwirkungen und Kontraindikationen zu überwachen…
Ganz herzlichen Dank für Ihre Einschätzung und Ihnen einen guten Start in die neue Woche!
Beste Grüße,
Marie K.
Sehr geehrte Marie K,
wenn die „Produkttester“ Personen sind, die von der Zweckbestimmung vorgesehen sind und das Produkt „sowieso“ anwenden, also ganz unabhängig davon, ob sie im Anschluss befragt werden, und(!) wenn diese Befragung keinen Einfluss auf die Anwendung des Produkts z.B. die Behandlungsdauer hat (beispielsweise weil die Patienten solange warten müssen), wäre das keine klinische Prüfung. Wenn eine der Bedingungen allerdings nicht erfüllt ist, wahrscheinlich schon.
Befragungen sind eine Möglichkeit, klinische Daten zu sammeln. Viele klinische Daten stammen nicht aus klinischen Prüfungen.
Viele Grüße
Christian Johner
Sehr geehrtes Team des Johner-Instituts,
mich beschäftigt eine Frage zur Interpretation von „Erfassung personenbezogener Daten“:
Wenn in der routinemäßigen Verwendung einer Medizinproduktesoftware Daten erhoben werden, dann sind sie natürlich personenbezogenen.
Für eine PMCF Analyse spielt der Personenbezug allerdings keine Rolle mehr, die Daten können vor der Analyse anonymisiert werden.
Sind dann die Forderungen, die für personenbezogene Daten gelten, überhaupt noch relevant?
Beste Grüße,
Peter Beck
Sehr geehrter Her Beck,
Daten, die anonymisiert sind, zählen nicht mehr als personenbezogene Daten. Damit sind auch die entsprechenden regulatorischen Anforderungen daran nicht mehr anwendbar.
Die Anonymisierung ist immer ein zweischneidiges Schwert. Einerseits hilft sie, regulatorischen Ärger zu vermeiden. Andererseits erschwert sie z.B. eine „Root Cause Analysis“ oder die Information gezielter Personen.
Danke für Ihre wichtige Frage!
Beste Grüße, Christian Johner
Sehr geehrtes Johner Team
Mit Interesse habe ich Ihren Artikel gelesen. Wir beginnen in unserer Firma wir eine retrospektive Kundenumfrage zur Wirksamkeit und Sicherheit bestimmter Medizinprodukte durchzuführen. Behandelnde Ärzte sollen einen kurzen Online-Fragebogen zur Wirksamkeit und Sicherheit dieser Medizinprodukte beantworten. Wir erheben keinerlei personenbezogenen Gesundheitsdaten von behandelten Patienten, sondern nur pauschale Erfahrungswerte der Anwender des Gerätes.
Wir diskutieren schon länger firmenintern, welche essenziellen Dokumente wir für diesen Survey erstellen müssen. Die vollständigen Anforderungen an die essenziellen Dokumente klinische Prüfungen nach ISO14155 zu erfüllen, scheint uns weit über das Ziel hinauszuschießen.
Gibt es Ihrerseits Erfahrungswerte bzw. Vorschläge? Dafür vielen Dank im Voraus.
Michael. F.
Sehr geehrter Herr F,
wenn Sie keine personenbezogenen Daten sammeln und diese Daten bei behandelnden Ärzten und nicht bei Patienten erheben (v.a. nicht in eine Behandlung eingreifen), dann sind weder der Datenschutz noch die Ethik ein Hindernis. Damit fallen Sie auch nicht unter den „Scope“ des Artikels, zu dem Sie die Frage gestellt haben. Sie benötigen keine Ethik-Kommission. Eine klinische Prüfung liegt auch nicht vor, so dass Sie auch nicht in den Scope der ISO 14155 fallen.
Insgesamt teile ich Ihre Einschätzung, dass bei dieser in jeder Hinsicht „harmlosen“ Sammlung von Daten nicht über das Ziel hinausgeschossen werden sollte. Vielmehr ist Ihr Bemühen anzuerkennen, Informationen aus dem Feld zu sammeln, um die Sicherheit, Leistungsfähigkeit und Wirksamkeit Ihrer Produkte zu sicherzustellen.
Viele Grüße
Christian Johner