Wie Sie die regulatorischen Anforderungen an die PMCF am schnellsten erfüllen
Die klinische Nachbeobachtung, auf Englisch Post-Market Clinical Follow-up, beanstanden die Benannten Stellen immer häufiger. Und das, nachdem die Hersteller die Hürde davor erfolgreich gemeistert haben: die initiale klinische Bewertung.
Dieser Artikel liefert Herstellern von Medizinprodukten
- eine schnelle Übersicht über die regulatorischen Anforderungen an den PMCF,
- eine Beschreibung der wichtigsten Methoden beim Post-Market Clinical Follow-up und
- Tipps, um die häufigsten Beanstandungen und damit unnötige Verzögerungen und Aufwände zu vermeiden.
1. PMCF: Die Grundlagen
a) Zielsetzung und Definition
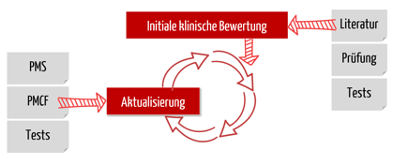
Die MDR verpflichtet die Hersteller dazu, im Rahmen der klinischen Bewertung die Sicherheit, die Leistungsfähigkeit und den Nutzen ihrer Medizinprodukte über den kompletten Produktlebenszyklus hinweg nachzuweisen. Für diese Nachweise sind auch klinische Daten erforderlich. Folglich müssen Hersteller diese klinischen Daten über den kompletten Produktlebenszyklus hinweg sammeln und bewerten.
Dieser kontinuierliche Prozess zur Aktualisierung der klinischen Bewertung (und damit der Aktualisierung der klinischen Daten) heißt Post-Market Clinical Follow-up. So definiert es auch der Teil B des Anhangs XVI der MDR:
“PMCF shall be understood to be a continuous process that updates the clinical evaluation referred to in Article 61 and Part A of this Annex and shall be addressed in the manufacturer’s post-market surveillance plan.”
MDR, Anhang XIV, Part B
Eine Bewertung, ob die ermittelten Risiken weiterhin vertretbar sind bzw. ob neue Risiken identifiziert wurden, ist Teil des PMCF.
b) Abgrenzung
Der PMCF ist eine Untermenge der Post-Market Surveillance (PMS). Die PMS beinhaltet das Sammeln aller Arten von Informationen aus der Praxis, die beispielsweise in Form von Serviceberichten, Anrufen bei der Hotline, Kundenbeschwerden usw. vorliegen. Die Ziele der PMS sind weitreichender als die Ziele des PMCF.
Eine genaue Abgrenzung von PMS und PMCF finden Sie in diesem Fachartikel zur PMS.
2. Regulatorische Anforderungen
a) Allgemeine Anforderungen
Viele Rechtssysteme fordern das kontinuierliche Sammeln und Bewerten klinischer Daten. Das Konzept des Post-Market Clinical Follow-up stammt jedoch von der MDR. Diese EU-Verordnung fordert in Artikel 10, Absatz 3, dass die klinische Bewertung eine klinische Nachbeobachtung von Medizinprodukten nach dem Inverkehrbringen umfasst – sprich: den Post-Market Clinical Follow-up.
In Anhang II, Absatz 6.1 (d) der MDR werden sowohl der Plan als auch der Bericht des Post-Market Clinical Follow-up (PMCF) zu den klinischen Daten gezählt. Die Dokumente gehören somit zur Technischen Dokumentation eines Medizinprodukts unter den Vorgaben der MDR.
b) Anforderungen an den Prozess
Die PMCF wird durchgeführt gemäß einer Methode, die in einem Plan für die klinische Nachbeobachtung nach dem Inverkehrbringen dargelegt ist. Die MDR fordert im Anhang XIV, Teil B, 6.1:
„Der Plan für die klinische Nachbeobachtung nach dem Inverkehrbringen beschreibt die Methoden und Verfahren für das proaktive Sammeln und Bewerten klinischer Daten, um
MDR, Anhang XIV, Teil B, 6.1
- die Sicherheit und die Leistung des Produkts während seiner erwarteten Lebensdauer zu bestätigen,
- zuvor unbekannte Nebenwirkungen zu ermitteln und die ermittelten Nebenwirkungen und Kontraindikationen zu überwachen,
- entstehende Risiken auf der Grundlage empirischer Belege zu ermitteln und zu untersuchen,
- die fortwährende Vertretbarkeit des Nutzen-Risiko-Verhältnisses gemäß Anhang I Abschnitte 1 und 9 zu gewährleisten und
- eine mögliche systematische fehlerhafte oder zulassungsüberschreitende Verwendung des Produkts festzustellen, damit überprüft werden kann, ob seine Zweckbestimmung angemessen ist.“
Die Benannte Stelle muss prüfen, ob der PMCF-Plan und seine Umsetzung angemessen sind.
Diese Überprüfung der klinischen Bewertung durch die Benannte Stelle umfasst auch die Verfahren des Herstellers und die Dokumentation der PMCF-Maßnahmen sowie ggf. die Begründung für die Nichtdurchführung der PMCF.
c) Anforderungen an die Dokumentation
Übersicht über die Dokumente
Benannte Stellen überprüfen in ihrem Clinical Evaluation Assessment Report (CEAR), ob ein PMCF-Plan aufgeführt und PMCF-Aktivitäten in einem PMCF-Bewertungsbericht dokumentiert sind:
“whether the post-market surveillance plan, including the PMCF plan, is adequate“
[…]
“Provide an assessment of the consistency of the clinical evidence with:
[…]
(b) the post-market clinical follow-up (PMCF) plan.”
MDCG-2020-13
Um diese Ziele zu erfüllen, stellt die Medical Device Coordination Group (MDCG) den Herstellern von Medizinprodukten eine wichtige Hilfestellung zur PMCF-Dokumentation zur Verfügung.
PMCF-Plan nach MDCG 2020-7
Das Template für einen Post-market clinical follow-up (PMCF) Plan – a guide for manufactures and notified bodies (MDCG 2020-07) umfasst 12 Seiten, die vom Hersteller produktspezifisch befüllt werden können. Dabei werden die Informationen stets im Tabellenformat abgefragt.

Abschnitt C listet die PMCF-Aktivitäten, die für das Medizinprodukt geplant sind. Dabei sollten Hersteller zunächst das Ziel der jeweiligen PMCF-Aktivität benennen und anschließend eine Begründung für deren Eignung angeben. Auch ist es erforderlich Fristen anzugeben, innerhalb derer die Aktivitäten durchzuführen sind. Dies Fristen sind vierteljährlich oder zumindest jährlich festzulegen.
Das Template nennt Beispiele für PMCF-Aktivitäten, die Folgendes umfassen:
- Medizinprodukteregister – Suche
- PMCF-Studie
- Nachweise aus der realen Welt („real-world evidence“)
- Umfragen
Zu jeder Aktivität ist hinzuzufügen,
- aus welchem Dokument die Forderung der geplanten PMCF-Aktivität kommt,
- eine Beschreibung der geplanten Aktivität (generelle Methode und Durchführung),
- das Ziel der PMCF-Aktivität,
- Methodik, die als Teil der Aktivität verwendet wird,
- Beschreibung der Gründe für die Angemessenheit der gewählten Methode sowie
- Frist und Timeline.
PMCF-Bewertungsbericht nach MDCG 2020-8
Der PMCF-Bewertungsbericht nach MDCG 2020-8 listet gemäß den Anforderungen der MDR die Ergebnisse der geplanten PMCF-Aktivitäten. Er ist ein Dokument der Technischen Dokumentation Ihres Medizinprodukts.
Die Schlussfolgerungen des PMCF-Bewertungsberichts werden berücksichtigt bei der Aktualisierung von:
- Klinischer Bewertung
- Risikomanagement-Akte
- PMCF-Plan
- Kurzbericht über Sicherheit und klinische Leistung (SSCP)
Insbesondere für Produkte der Klasse III und implantierbare Produkte wird der PMCF-Bewertungsbericht mindestens einmal jährlich anhand dieser Daten aktualisiert. Der PMCF-Bewertungsbericht ist Teil des klinischen Bewertungsberichts und der Technischen Dokumentation.
d) Ausnahmen
Nicht in allen Fällen müssen Aktivitäten im Rahmen des Post-Market Clinical Follow-ups durchgeführt werden. Die MDR schreibt hierzu:
“(b) The post-market surveillance plan shall cover at least:
— a PMCF plan as referred to in Part B of Annex XIV, or a justification as to why a PMCF is not applicable.”
MDR, Anhang III, 1.1. b)
Ein großes Problem ist, dass es weder Hinweise gibt, bei welchen Produktgruppen oder Klassifizierungen dieses angewendet werden kann, noch Formulierungsbeispiele, die für eine Begründung herangezogen werden können.
Benannte Stellen überprüfen in ihrem Clinical Evaluation Assessment Report (CEAR), ob eine Begründung angeben und adäquat ist:
“If no PMCF is planned, has the manufacturer provided an acceptable justification for not conducting a PMCF?”
MDCG-2020-13
Es gibt Medizinprodukte, die keine medizinische Zweckbestimmung haben oder bei denen das Sammeln klinischer Daten nicht sinnvoll ist. Beispiele dafür sind Zubehör und einige Medizinprodukte, z. B. Steckbeckenspülapparate zum Reinigen von Bettpfannen oder Turbinen für Dentalbohrer.
In diesen Fällen, in denen klinische Daten ungeeignet sind, um die Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen nachzuweisen, wird eine klinische Bewertung auf Basis des Artikel 61 (10) durchgeführt. Dann kann der Hersteller auf andere Nachweise wie nichtklinische Testmethoden einschließlich Leistungsbewertung und technischer Prüfung („bench testing“) zurückgreifen. Da es keine klinisch relevanten Endpunkte gibt, müssen keine PMCF-Daten erhoben werden.
Wie können Sie feststellen, ob und in welchen Umfang PMCF-Aktivitäten zweckmäßig sind? Gehen Sie wie bei einer klinischen Bewertung zur klinischen Evidenz an die Sache und betrachten Sie die Art, die Klassifizierung, die Zweckbestimmung sowie Risiken des Produkts.
Das bestätigt auch die MDR:
“That level of clinical evidence shall be appropriate in view of the characteristics of the device and its intended purpose.”
MDR, Artikel 61, 1
“The clinical evaluation shall be thorough and objective, and take into account both favourable and unfavourable data. Its depth and extent shall be proportionate and appropriate to the nature, classification, intended purpose and risks of the device in question, as well as to the manufacturer’s claims in respect of the device.”
MDR, Anhang XIV, 2
Um zu begründen, warum keine PMCF-Aktivtäten notwendig sind, nutzen Sie die Formulierungen oder Betrachtungen
- aus dem MDCG 2020-6 zur Beschreibung von bewährter Technik (well-established technology),
- auf der Grundlage des Risikomanagements und unter Berücksichtigung der besonderen Merkmale des Zusammenspiels zwischen dem Produkt und dem menschlichen Körper, der bezweckten klinischen Leistung und der Angaben (claims) zum Produkt.
Beachten Sie den Fachartikel zur MDCG 2020-6.
3. Arten von PMCF-Aktivtäten
a) Übersicht und Minimalanforderung
Die MDR unterscheidet bei den PMCF-Aktivitäten die allgemeinen Methoden und Verfahren der PMCF sowie spezifische Methoden und Verfahren der PMCF:
(a) the general methods and procedures of the PMCF to be applied, such as gathering of clinical experience gained, feedback from users, screening of scientific literature and of other sources of clinical data;
(b) the specific methods and procedures of PMCF to be applied, such as evaluation of suitable registers or PMCF studies.
MDR, Annex XIV, Part B 6.2
Als Minimum müssen produktspezifische Literatursuchen durchgeführt werden, um sogenannte pivotale Daten aus in nach dem Peer-Review-Verfahren überprüfter wissenschaftlicher Fachliteratur veröffentlichten Berichten zu erhalten.
Der Fachartikel zur systematischen Literaturrecherche gibt Ihnen konkrete Hilfestellungen.
Bei implantierbaren Produkten und Produkten der Klasse III, bei denen keine klinischen Prüfungen gemäß Artikel 61 Absatz 4 durchgeführt wurden, umfasst der PMCF-Plan Studien, die nach dem Inverkehrbringen der Bestätigung der Sicherheit und Leistung des Produkts dienen.
b) Zusätzliche PMCF-Aktivitäten
Übersicht
Weiterhin gehören folgende Aktivitäten zum Post-Market Clinical Follow-up:
| PMCF-Aktivität | Beschreibung | Beispiele |
| Post-Market Studien | PMCF-Studien sind klinische Prüfungen im Sinne des Artikels 74 MDR, die nach der Inverkehrbringung des Produkts durchgeführt werden. Zu diesem Zeitpunkt trägt das Produkt bereits eine CE-Kennzeichnung. | |
| Erfassung von Daten in Registern | Ein Hersteller-Produktregister, das spezifisch ist für die Art des Produkts oder die Gruppe der Medizinprodukte, zu der das Produkt gehört. Es ist im Voraus festzulegen, welche qualitativen und quantitativen Daten – auf der Grundlage des Risikos des Produkts und des zugehörigen Zubehörs – gesammelt und analysiert werden sollen. | |
| Systematische Befragung von Patienten/Nutzern – Anwenderbefragungen (Survey) | Geplante Erhebungen zur Sammlung von Informationen über die Verwendung des betreffenden Medizinprodukts | |
| Publikationen | Frei verfügbare Dokumentation zum eigenen Produkt | |
| Real-world Evidence Analyses (RWE-Analysen) | Die Daten fallen automatisch an und eine Planung ist abhängig von Zielen und im Sinne einer optimalen Nutzung der Daten zu empfehlen. Es liegt hier keine regulatorische Anforderung vor; es werden keine konkreten Endpunkte verfolgt; es gibt keine zeitliche Beschränkung und keine Fallzahlplanung. |
Genauere Anleitungen zu den PMCF-Studien finden Sie im entsprechenden Fachartikel.
Klinische Prüfungen
Klinische Prüfungen sind meist konfirmatorischer Natur und setzten eine aufwendige Studienplanung sowie eine klare Beschreibung der Studienbedingungen voraus. Dabei kann jeder Störfaktor zu einer erhöhten Streuung führen und somit erheblichen Einfluss auf die statistisch notwendige Fallzahl haben.
So ist es nicht verwunderlich, dass über geeignete Ein- und Ausschlusskriterien die Studienpopulation möglichst homogen gehalten werden soll. Das spart Zeit und Kosten, führt aber auch zu Idealbedingungen, unter denen die gewünschten klinischen Endpunkte eines Medizinprodukts überprüft werden. Inwieweit die dabei erzeugten klinischen Daten noch die routinemäßige Anwendung repräsentieren, mag bei der ein oder anderen Studie durchaus angezweifelt werden.
Im Gegensatz zu klinischen Prüfungen kann der Einsatz von Medizinprodukten auch im Versorgungsalltag erprobt werden. Die Studienbedingungen und besonders die Auswahl der Teilnehmer werden dabei nicht über Ein- und Ausschlusskriterien gesteuert. Das Ergebnis solcher pragmatischen klinischen Studien (PCT, Pragmatic Clincial Trials) sind Real World Data (RWD), mit denen die Lücken zwischen der experimentellen und der routinemäßigen Anwendung eines Medizinprodukts geschlossen werden können.
4. Häufige Fehler
a) Mangel an produktspezifischen klinischen Daten
Äquivalenzbetrachtung unter der MDR nicht mehr möglich
Die bisherige Strategie klinischer Bewertungen unter der MDD umfasste zum Großteil den Nachweis über klinische Daten zu Äquivalenzprodukten. Die Anforderungen zum Nachweis an die Äquivalenz sind unter der MDR gestiegen.
Beachten Sie den Artikel zur Äquivalenz und zur MDCG 2020-5.
Im Nachgang der Zulassung wurde jedoch die Erhebung weitere klinischer Daten zum eigentlichen Produkt vernachlässigt. Dies führt dazu, dass
- Langzeitauswirkungen von Produkten nicht erhoben und ausgewertet wurden,
- nicht betrachtete Risiken unentdeckt blieben sowie
- Erweiterungen der klinischen Anwendungsgebiete nicht berücksichtigt wurden.
Forderung von PMCF-Studien zur MDR-Zertifizierung
Mit der expliziten Forderung nach der proaktiven Erhebung von klinischen Daten im PMCF-Prozess sollen diese Themen adressiert werden. Sie dienen dazu, die Sicherheit der Medizinprodukte zu verbessern und ihre vorhergesehene Leistung stetig zu überprüfen.
Im Allgemeinen müssen ausreichende klinische Nachweise vorliegen, um die Sicherheit, die Leistung und die Annehmbarkeit der Nutzen-Risiko-Bestimmung in Bezug auf den Stand der Technik für die Altprodukte vor der CE-Kennzeichnung gemäß der MDR zu bestätigen. Ggf. müssen sogar PMCF-Studien durchgeführt werden, um den Mangel an klinischen Daten zu beheben.
Das MDCG 2020-6 sieht bei fehlender Äquivalenz und bei produktspezifischen Risiken PMCF-Studien vor:
„The European Commission guidance MEDDEV 2.12/2 regarding PMCF studies notes different instances where a PMCF study may have been justified:
– Route chosen for clinical evaluation: where CE marking for legacy devices was based upon equivalence, PMCF studies may have been necessary. The European Commission guidance MEDDEV 2.12/2 regarding PMCF also notes that in the case that clinical evaluation was based exclusively on clinical data from equivalent devices for initial conformity assessment, the certifying notified body shall verify that PMCF studies have been conducted, in accordance with the relevant provisions of the Directives.
– Device related factors: There are a number of device-related factors where PMCF studies may have been necessary.
When assessing the conformity of legacy devices under the MDR, it is important to verify whether PMCF studies considered necessary under the MDD/AIMDD (and where applicable, during the transition period, under the MDR), have been appropriately conducted, and results are taken fully into account for in the clinical evaluation for the conformity assessment under MDR.“
MDCG 2020-6
Fazit: Ein solcher Nachweis sollte sich nicht auf neue PMCF-Studien stützen, die im Rahmen der MDR begonnen wurden, um Lücken zu schließen (z. B. Indikationen, die nicht durch klinische Nachweise belegt sind).
Weiterführende Informationen finden Sie im Fachartikel zur MDCG 2020-6.
b) Keine Berücksichtigung von Off-Label-Daten
Off-Label Use zur Indikationserweiterung
Hersteller stellen sich oft die Frage, ob Off-Label-Daten verwendet werden können, um den beabsichtigten Zweck oder die beabsichtigten Indikationen zu erweitern. Wie mit diesen Daten umgegangen werden kann, beschreibt das Positionspapier des Team-NB.
Unter ‚Off-Label Use‘ eines Medizinprodukts versteht man im Allgemeinen, wenn ein Produkt außerhalb der Zulassung eingesetzt wird, also nicht wie in der Gebrauchsanweisung beschrieben:
- Außerhalb bestimmter Patientengruppen, z. B. in der Pädiatrie
- Für ein anderes Stadium oder einen anderen Schweregrad der Krankheit
- Für einen ähnlichen (nicht identischen) klinischen Zustand
- Einbringung in den Körper über andere Wege
Der Begriff ‚Off-Label Use‘ wird in der MDR in Anhang XIV, Teil B in dem Zusammenhang erwähnt, dass der PMCF-Plan des Herstellers eine systematische Fehlanwendung oder einen Off-Label-Gebrauch des Produkts ermitteln muss, um zu überprüfen, ob die Zweckbestimmung des Produkts korrekt ist. Jedoch werden ‚Off-Label Use‘ oder ‚Missbrauch‘ in der MDR nicht definiert.
Off-Label-Daten können zwar quantitativ ausreichend sein, insbesondere wenn ein systematischer Off-Label Use festgestellt wurde, aber sie haben oft keine ausreichende Qualität im Sinne aussagekräftiger Schlussfolgerungen. Off-Label-Daten werden in der Regel außerhalb formeller Protokolle erhoben, und das Fehlen von Protokollen führt letztlich dazu, dass die Qualität nicht ausreicht und keine evidenzbasierten Schlussfolgerungen gezogen werden können.
‚Foreseeable Misuse‘, ‚unsystematischer Misuse‘ und ’systematischer Misuse‘
Hersteller müssen vorhersehbare Fehlanwendungen durch Gebrauchstauglichkeitsstudien oder Berichte über klinische Prüfungen vor dem Inverkehrbringen identifizieren. Aber es ist oft schwierig, Bereiche künftiger Fehlanwendungen vorherzusagen, und ohne die direkte Aufsicht des Herstellers über die Verwendung jedes einzelnen Produkts ist es unvermeidlich, dass es zu einer nicht bestimmungsgemäßen Verwendung kommt. Wenn eine missbräuchliche Verwendung festgestellt wird, muss der Hersteller die Risiken gemäß den Risikokontrollmaßnahmen beseitigen oder kontrollieren.
Beachten Sie auch die Fachartikel zu den Usability-Studien und zum Risikomanagement.
Der Hersteller sollte beim dokumentierten Off-Label Use zwischen systematischem Misuse und unsystematischem Misuse differenzieren:
| Unsystematischer Misuse | Systematischer Misuse | |
| Details | Bei nicht gedecktem medizinischem Bedarf („unmet medical need“) und wenn keine anderen zugelassenen praktikablen Alternativen zur Verfügung stehen, könnte es für einen Arzt ethisch vertretbar sein, alternative Optionen in Betracht zu ziehen. Ein Produkt wurde „unsystematisch“, d. h. zufällig, verwendet. | Ein Produkt wird wiederholt oder kontinuierlich außerhalb seiner genehmigten Zweckbestimmung und Indikationen verwendet. |
| Quellen | Über eine solche Verwendung eines Produkts wird in der Fachliteratur häufig in Form von Einzelfallstudien berichtet. Hersteller können eine solche Off-Label-Verwendung im Rahmen ihrer allgemeinen Aktivitäten nach der Markteinführung durch Literaturrecherchen ermitteln. | Literatur, Anwenderbefragung |
| Klinische Daten | ja | ja |
| Betrachtung in klinischer Bewertung | ja | ja |
| Dokumentation im PMCF-Bericht | nein | ja |
| Erweiterung der Zweckbestimmung | nein | nein |
Wenn Hersteller einen systematischen Off-Label Use feststellen, sollten sie geeignete Maßnahmen ergreifen, um nicht nur den Fehlgebrauch einzudämmen, sondern auch um zu prüfen, ob in der medizinischen Fachwelt ein echter Bedarf für die neu festgestellte Anwendung in Bezug auf den spezifischen medizinischen Zweck/die Indikation besteht.
c) Zeiträume der Dokumentation und Durchführung der Maßnahmen sind nicht angemessen
Die MDR gibt Zeiträume für die Erstellung des PSUR vor, der die wichtigsten Ergebnisse des PMCF-Prozesses enthalten muss. Das heißt aber nicht, dass die Durchführung und Auswertung von PMCF-Aktivitäten daran gekoppelt sind.
Es gibt Aktivitäten, die häufiger durchgeführt werden sollten, z. B. eine Literaturrecherche. Weiterhin geben Interimsanalysen von PMCF-Studien oder Anwenderbefragungen Hinweise, um geeignete Maßnahmen bereits abzuleiten oder Anpassungen am Studienprotokoll oder -design vorzunehmen, z. B. bei Änderungen der Patientenzahl oder von Studienzentren.
Die MDR unterscheidet bei der Dokumentation der PMCF-Aktivitäten hinsichtlich der Klassifizierung von Produkten:
For class III devices and implantable devices, the PMCF evaluation report and, if indicated, the summary of safety and clinical performance referred to in Article 32 shall be updated at least annually with such data.
MDR, Artikel 61, 11
Im PSUR ist jeweils eine Zusammenfassung der wichtigsten Ergebnisse des PMCF-Bewertungsberichts gefordert. Das bedeutet, dass der PMCF-Bewertungsbericht eine ausführliche Analyse und Bewertung der PMCF-Daten enthält (MDCG-Dokument 2020-8) und „nur“ die wesentlichen Punkte in den PSUR übernommen werden (MDCG-Dokument 2022-21).
Nun kann es sein, dass während eines zweijährigen Zeitraums zur PSUR-Erstellung mehrere PMCF-Bewertungsberichte erstellt werden, weil das Intervall dafür kürzer ist oder es mehrere Aktivitäten mit unterschiedlichen Intervallen gibt. Es ist zu empfehlen, von jedem PMCF-Bewertungsbericht eine kurze Zusammenfassung im PSUR aufzuführen.
Vor jeder Erstellung des PSUR ist es auf jeden Fall angebracht, alle angefallenen PMCF-Analysen und -Bewertungsberichte aus dem zu bewertenden Zeitraum durchzugehen und zu schauen, ob alles erledigt ist. Sollten PMCF-Aktivtäten noch nicht abgeschlossen sein, muss im PSUR begründet werden, warum es evtl. eine Abweichung vom Plan gab oder noch keine ausreichenden PMCF-Ergebnisse vorliegen.
Weitere Hinweise enthält der Artikel zum PMS-Bericht bzw. PSUR.
5. Fazit und Unterstützung
Medizinprodukte müssen über den kompletten Lebenszyklus sicher, leistungsfähig und wirksam sein. PMCF-Aktivitäten sind eine von der MDR geforderten Maßnahme und die weiterführende Methode, um diesen Nachweis zu führen. Das heißt aber nicht, dass PMCF-Prozesse oder PMCF-Studien immer zwingend notwendig sind.
Der Umfang sollte angemessen sein in Bezug auf Art, Klassifizierung, Zweckbestimmung und Risiken des betreffenden Produkts.
Im Rahmen der klinischen Bewertung werden Lücken in klinischen Daten aufgedeckt und initial proaktive PMCF-Aktivitäten festgelegt.
Holen Sie sich die passende Unterstützung bei der Erstellung Ihrer gesamten klinischen Bewertungsakte sowie bei der Konzeption, Durchführung und Auswertung Ihrer PMCF-Aktivitäten. Das Johner Institut bietet Ihnen:
- Outsourcing Ihrer klinischen Bewertung und Planung Ihrer klinischen Datenerhebung
- Seminare zur klinischen Bewertung zum Erwerben der notwendigen Kompetenzen
Melden Sie sich gleich mit einem Terminvorschlag für ein kostenfreies und unverbindliches Gespräch.
Nehmen Sie Kontakt auf mit den Expert:innen des Johner Institut. Diese prüfen Ihre klinische Bewertungsakte und PMCF-Dokumente und geben Ihnen konkrete Hinweise zur Verbesserung. Sie unterstützen Sie auch bei der Fallzahlplanung und Auswertung Ihrer PMCF-Daten.