Der Begriff ‚Predicate Device‘ fällt meist im Kontext von 510(k)-Zulassungen der FDA. Allerdings definiert die FDA diesen Begriff nicht. Sie legt hingegen fest, was ‚Substantial Equivalence‘ ist. Klingt kompliziert?
Der Nachweis der Äquivalenz ist keinesfalls nur im Kontext der FDA relevant. Deshalb verschafft dieser Artikel Klarheit – gerade, aber nicht nur für Hersteller, die ihre Medizinprodukte in den USA vermarkten wollen.
1. Predicate Device: Die Grundlagen
a) Was ist ein Predicate Device?
‚Predicate Device‘ ist kein definierter Begriff. Der FD&C-Act in Section 513(i) definiert den Begriff ‚Substantial Equivalence‘:
(1) (A) For purposes of determinations of substantial equivalence under subsection (f) and section 360j(l) of this title, the term „substantially equivalent“ or „substantial equivalence“ means, with respect to a device being compared to a predicate device, that the device has the same intended use as the predicate device and that the Secretary by order has found that the device has […]”.
FD&C Section 513(i)
Die vollständige Definition findet sich weiter unten.
Demnach dient das Predicate Device als das Vergleichsprodukt, mit dem die Äquivalenz zu einem Produkt, das ein Hersteller in den USA vermarkten möchte, nachgewiesen wird. Der Hersteller erbringt den Nachweis selbst, und die FDA überprüft diese ‚Substantial Equivalence‘ im Rahmen der Premarket Notification, besser bekannt als das 510(k)-Verfahren.
Gemäß 21 CFR 807.92 (a) (3) kann als Vergleichsprodukt dienen:
- Ein Medizinprodukt, das von dem 28.05.1976 legal in den USA vermarktet wurde (d. h. vor Einführung der risikobasierten Klassifizierung): ein ‚Pre-Amendment Device‘
- Ein in den USA legal vermarktetes Medizinprodukt, das die FDA der Klasse I oder II zugewiesen hat
- Ein in den USA legal vermarktetes Medizinprodukt, für das bereits über ein 510(k)-Verfahren die Substantial Equivalence nachgewiesen wurde
b) Wann bzw. warum muss man das bestimmen?
Ein Predicate Device wird im 510(k)-Verfahren benötigt. Dies ist das übliche Zulassungsverfahren in den USA für Produkte der Klasse II sowie für bestimmte Produkte der Klassen I und III. Allerdings sind einige Klasse-II-Produkte von der Premarket Notification befreit (‚Exempt‘-Status).
Eine Liste der ‚Exempt Devices‘ führt die FDA auf ihrer Webseite.
Im 510(k)-Verfahren reicht es aus, wenn der Hersteller die Äquivalenz mit einem Predicate Device nachweist. Gelingt dies, geht die FDA davon aus, dass das Produkt mindestens genauso sicher und wirksam ist und erteilt die Marktfreigabe (510(k) Clearance).
Ohne den Nachweis der Äquivalenz muss ein sehr aufwendiges De-Novo– oder gar PMA-Verfahren durchlaufen werden. Dieses führt zu höheren Kosten, höheren Anforderungen an die Nachweise (z. B. klinische Daten) und eine längere Zulassungsdauer.
c) Was ist der Unterschied zu den gleichartigen Produkten (MDR)?
Rolle der Äquivalenzbetrachtung
Die MDR kennt ein ähnliches Konzept. Sie spricht in der englischen Version von ‚Equivalent Device‘, in der deutschen Übersetzung von „gleichartigen“ Produkten (siehe Artikel 61 MDR).
Dieser Vergleich mit einem gleichartigen Produkt ist allerdings beschränkt auf die klinische Bewertung und nicht auf das gesamte Konformitätsbewertungsverfahren.
Durch einen Nachweis der Gleichartigkeit ist es unter der MDR möglich, klinische Daten von eben jenem Vergleichsprodukt für die eigene klinische Bewertung heranzuziehen. In bestimmten Fällen kann dadurch sogar auf eine klinische Prüfung von Hochrisiko-Produkten verzichtet werden (Artikel 61 (4) MDR).
Bewertungskriterien
Unterschiedlich sind auch die Bewertungskriterien bezüglich der Äquivalenz bzw. Gleichartigkeit zwischen FDA und MDR:
- Die MDR unterscheidet nach technisch, biologisch und klinisch.
- Die FDA unterscheidet nach ‚Intended Use‘ und ‚Technological Characteristics‘.
Man kann sagen, dass die MDR in Zusammenhang mit der MEDDEV 2.7/1 rev.4 strenger ist, was den Vergleich der Gleichartigkeit angeht. So wird unter der MDR z. B. der Einsatz von gleichen Materialien verlangt oder identische Indikationen und Körperregionen. Hier ist die FDA weniger streng.
Es gibt erfolgreiche 510k-Verfahren, in denen die Äquivalenz einer Software zur Beurteilung von Anomalien anhand von Bilddaten mit einer Software für die Beurteilung und Charakterisierung von Anomalien in der Brust anhand von MR-Bilddaten erfolgreich nachgewiesen und von der FDA zugelassen wurde (Quelle).
2. Ein Predicate Device bestimmen
a) Definition und Algorithmus
Section 513(i) des FD&C Acts bestimmt, wann zwei Produkte als „substantially equivalent“ betrachtet werden dürfen:
“’substantial equivalence‘ means, with respect to a device being compared to a predicate device, that the device has the same intended use as the predicate device and that the Secretary by order has found that the device-
(i) has the same technological characteristics as the predicate device, or
(ii) (I) has different technological characteristics and the information submitted that the device is substantially equivalent to the predicate device contains information, including appropriate clinical or scientific data if deemed necessary by the Secretary or a person accredited under section 360m of this title, that demonstrates that the device is as safe and effective as a legally marketed device, and (II) does not raise different questions of safety and effectiveness than the predicate device.”
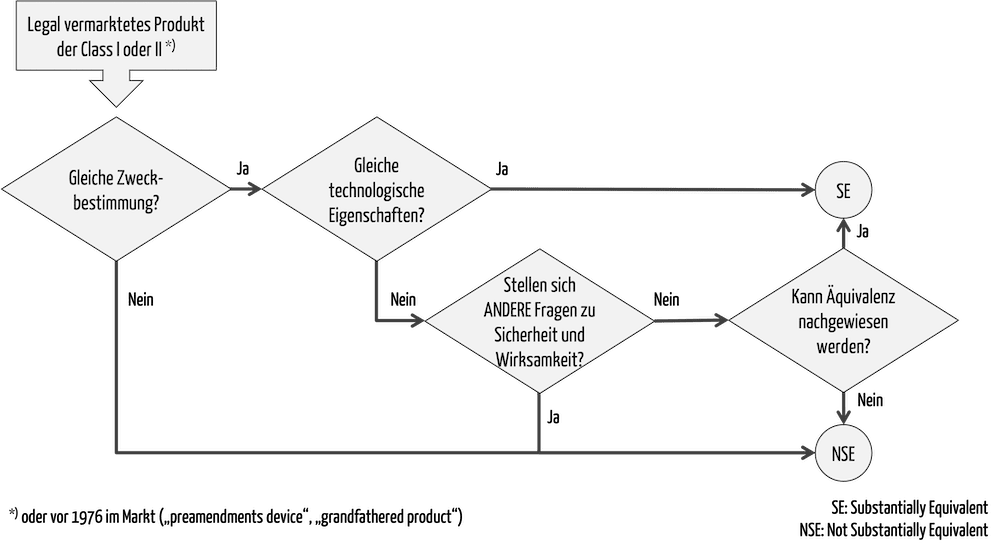
Ob es für ein Produkt ein Predicate Device gibt, also ob das zweite Produkt “substantially equivalent” im Sinne der FDA ist, lässt sich mit einem Entscheidungsbaum feststellen (Abb. 1).
Pflichtlektüre sind unter anderem diese drei Guidance-Dokumente der FDA:
- The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] (veröffentlicht 2014)
- Benefit-Risk Factors to Consider When Determining Substantial Equivalence in Premarket Notifications (510(k)) with Different Technological Characteristics (veröffentlicht 2018)
- Best Practices for Selecting a Predicate Device to Support a Premarket Notification [510(k)] Submission (Entwurf vom 07.09.2023)
b) Kriterium 1: Gleiche Zweckbestimmung
Zunächst müssen Hersteller nachweisen, dass die Zweckbestimmung identisch ist mit der des Predicate Device. Im Gegensatz zu den Kriterien der MDR im Rahmen der klinischen Bewertung meint die FDA hier allerdings den übergeordneten medizinischen Zweck. Unterschiede bei Indikationen, Patientenpopulation oder Anwendern führen demnach nicht zwingend zu einer neuen Zweckbestimmung.
Im Beispiel der Software zur Beurteilung von Anomalien anhand von Bilddaten ist der Hersteller der Ansicht, dass eine unterschiedliche Körperregion und Indikation nicht zu einer anderen, übergeordneten Zweckbestimmung führen, nämlich der Beurteilung bestimmter Krankheitszustände mithilfe von Standard-Scoring unter Verwendung von Algorithmen des maschinellen Lernens. Diesem Vergleich hat die FDA positiv zugestimmt.
Lesen Sie in diesem Artikel mehr zu den Inhalten einer Zweckbestimmung. Beachten Sie: In der Abb. 1 ist die unterste Einheit der medizinische Zweck. Die FDA unterscheidet hier genauer.
c) Kriterium 2: Technologische Eigenschaften
Identische technologische Eigenschaften wird es selten geben. Ansonsten würden wir auf einem alten Stand der Technik stehen bleiben. Wenn die FDA von technologischen Eigenschaften spricht, meint sie Materialien, Energiequellen, Algorithmen oder das Design im Allgemeinen.
Auch wenn diese nicht identisch sind, ist eine 510(k)-Clearance möglich. Allerdings dürfen die Unterschiede in der Technologie keine anderen (!) Fragestellungen bezüglich Sicherheit oder Wirksamkeit aufwerfen.
Mit “andere” bezieht sich die FDA insbesondere auf Fragen zur Sicherheit und Wirksamkeit, die sich beim Predicate Device nicht gestellt haben, beim vorliegenden Produkt allerdings zu Bedenken hinsichtlich Sicherheit oder Wirksamkeit führen.
Beispiel 1: Ein Stirnthermometer ist kein geeignetes Predicate Device für ein Ohrthermometer. Beim Ohrthermometer stellen sich Fragen bzw. existieren Risiken zur Biokompatibilität. Dies ist beim Stirnthermometer, das ohne Kontakt zum Patienten die Körpertemperatur erfasst, nicht der Fall.
Beispiel 2: Ein Oropharyngealtubus zum Offenhalten der Atemwege ist kein geeignetes Predicate Device für ein nicht-invasives Gerät, das die Atemwege offen hält, indem von außen auf den Hals ein Vakuum angewandt wird. Bei letzterem wird ein kontinuierlicher Druck auf Gewebe und Nervenstrukturen appliziert. Das wirft Fragen auf zu Risiken in Verbindung mit der Stimulation der Nerven.
d) Kriterium 3: Anwendung von Best Practices
Die FDA möchte ihr in die Jahre gekommenes 510(k)-Verfahren modernisieren. Dazu beschreibt sie weitere Kriterien für die Auswahl eines geeigneten Predicate Device.
Zukünftig möchte die FDA besser verstehen, warum Hersteller von einer Auswahl an validen Predicate Devices einem bestimmten Predicate Device Vorrang geben. Hersteller müssen ihre Wahl begründen.
Folgende Kriterien sollten Einfluss auf die Auswahl haben:
- Anwendung bewährter Methoden
Das Predicate Device sollte unter Anwendung von bewährten Methoden von der FDA freigegeben worden sein. Dazu zählen Vorgaben aus aktuellen anerkannten Normen (recognized consensus standards), aktuellen Guidance-Dokumenten der FDA oder die Nutzung von Medical Device Development Tools (MDDTs). Dies führt indirekt dazu, dass Hersteller ihre Produkte zukünftig nicht mehr mit (sehr) alten Predicate Devices vergleichen können. - Kontinuierliche Erfüllung der Sicherheits- und Leistungsanforderungen
Im besten Fall erreichen oder übertreffen Medizinprodukte über ihre Lebensdauer das Maß an Sicherheit und Leistung, das initial zur Zulassung bestimmt wurde. Somit sind keine Produkte als Predicate Device geeignet, zu denen es beispielsweise vermehrt Meldungen zu unerwünschten Ereignissen oder Vorkommnissen gibt (Tod, Verletzung, Fehlfunktionen), die ursprünglich nicht bedacht wurden, oder eine Erhöhung in der Häufigkeit oder dem Schweregrad der Vorkommnisse. Dies könnte beispielsweise auf grundlegende Probleme mit dem Design hindeuten. - Ungelöste Benutzungs- oder designbezogene Sicherheitsprobleme
Bekannt gewordene Sicherheitsprobleme des Predicate Device, z. B. durch mangelnde Gebrauchstauglichkeit oder problematisches Design, sollten mitigiert worden sein. - Designbezogene Rückrufe
Das Predicate Device sollte nicht aufgrund von designbezogenen Mängeln vom Markt zurückgerufen worden sein.
3. Ein Predicate Device finden
Schritt 1: Suche in FDA-Datenbank
Ein guter erster Schritt besteht darin, in der FDA 510(k)-Datenbank nach bekannten ähnlichen Produkten (z. B. der Konkurrenz) zu suchen (über Handelsnamen, Herstellernamen oder 510(k)-Nummer).
Schritt 2: Suche in Klassifizierungsdatenbank
Wird man hier nicht fündig oder gibt es keine bekannten ähnlichen Produkte, kann der Weg über die Klassifizierungsdatenbank erfolgreich sein. Hier wählt man zunächst die richtige Produktkategorie (“product code”) und kann dann in der 510(k)-Datenbank nach zugelassenen Produkten suchen.
Schritt 3: Prüfung anhand der Kriterien
Hat man auf diesem Wege mögliche Predicate Devices ausgewählt, müssen diese gegen die oben genannten Kriterien geprüft werden. Dazu tragen die Hersteller die folgenden Informationen zusammen:
- Zweckbestimmung
Hier lohnt sich generell ein Blick in die öffentliche Zusammenfassung der 510(k), die „510(k) Summary“. Auch ein Blick in die Gebrauchsanweisung (soweit verfügbar) oder auf die Webseite des Herstellers sind hilfreich. - Technologische Eigenschaften
Informationen über grundlegende technologische Eigenschaften finden sich in der 510(k) Summary, in der Gebrauchsanweisung oder über sonstige öffentlich zugängliche Informationen. Über den Freedom of Information Act (FoIA) ist es möglich, bei der FDA vollständige 510(k)-Akten anzufordern. Dieser Prozess kann allerdings auch mal über ein Jahr dauern; zudem sind vertrauliche Informationen in der Regel geschwärzt. - Zusätzliche Kriterien
- Bewährte Methoden: Diese sollten in der 510(k) Summary erwähnt worden sein. Manchmal finden sich in der Gebrauchsanweisung oder der Kennzeichnung auch Hinweise auf angewandte Normen.
- Post-Market Informationen: Informationen zu Sicherheitsproblemen und Rückrufen sind öffentlich über verschiedene FDA-Datenbanken zugänglich:
4. FAQ zu Predicate Devices
Frage 1: Muss es genau ein Predicate Device sein?
Im Idealfall reicht ein Predicate Device für den Nachweis der Substantial Equivalence. Manchmal lässt sich solch ein Produkt aber nicht finden. Dann ist es auch möglich, zwei oder mehrere Produkte als Predicate Device heranzuziehen. Die FDA empfiehlt aber, das ähnlichste Produkt als das ‚Primary Predicate Device‘ zu kennzeichnen.
Der Nachweis der Substantial Equivalence muss gegen alle gewählten Predicate Devices erfolgen. Das heißt: Die übergeordnete Zweckbestimmung muss mit allen Predicate Devices identisch sein. Technologisch darf es, wie oben erläutert, Unterschiede geben.
Beispiel Vitalmonitor: Für einen Monitor zur Überwachung von Vitalparametern wäre es denkbar, einzelne Predicate Devices für spezifische Vitalparameter (z. B. Körpertemperatur, Blutdruck, Herzfrequenz) auszuwählen.
Frage 2: Was ist der Unterschied zu einem ‚Reference Device‘ (FDA)?
Die FDA kennt neben den Predicate Devices sogenannte ‚Reference Devices‘. Diese können eine 510(k)-Einreichung bzw. den Nachweis der Substantial Equivalence unterstützen. Sie ersetzen aber kein Predicate Device.
Mit einem Reference Device möchten Hersteller die FDA beispielsweise auf Produkte hinweisen, die zwar eine andere Zweckbestimmung haben und somit nicht als Predicate Device geeignet sind, aber ähnliche technologische Eigenschaften (z. B. Materialien) in einem anderen Kontext nutzen und erfolgreich zugelassen wurden.
Das Reference Device unterstützt im Entscheidungsbaum (Abb. 1) die letzte Frage nach dem Nachweis der Äquivalenz (z. B. bezüglich der Testmethode).
Frage 3: Wie dokumentiert man die Äquivalenz?
Als Kernelement einer 510(k)-Akte dient eine Vergleichstabelle zwischen dem neuen Produkt und dem Predicate Device. In dieser Tabelle sollte der Hersteller Gemeinsamkeiten und Unterschiede bezüglich Zweckbestimmung, Indikationen sowie relevanten technologischen Eigenschaften detailliert auflisten und diskutieren. Zu jedem Eintrag sollte es eine Bewertung der Äquivalenz geben (z. B. “same”, “similar”, “different”).
Bei Unterschieden sollte genau begründet werden, warum diese nicht zu anderen Fragestellungen bezüglich der Substantial Equivalence führen. Hersteller sollten hier möglichst konkret werden: Beispielsweise können die Anwendung gleicher Methoden (z. B. gleiche Norm) und die Erfüllung gleicher Anforderungen (z. B. Grenzwerte wie in der Norm genannt) zusammen mit den entsprechenden Nachweisen als Begründung dienen.
Die Äquivalenz untermauern Hersteller durch weitere Informationen zu den angewandten Methoden (siehe oben) und die zugehörigen Nachweise. Generell akzeptiert die FDA einen least burdensome approach. Das heißt beispielsweise, dass sie in bestimmten Fällen auf die Einreichung von vollständigen Prüfberichten verzichtet.
5. Tipps
Tipp 1: Zweckbestimmung nicht splitten
Da die FDA explizit mehrere Predicate Devices erlaubt, liegt die Versuchung nahe, die Zweckbestimmung auf mehrere Predicate Devices zu splitten. Das wird dazu führen, dass die FDA die Argumentation ablehnt, dass das Produkt ’substantialy equivalent‘ sei. Die Zweckbestimmung muss über alle Predicate Devices hinweg gleich sein.
Tipp 2: Pre-Submission-Meeting nutzen
Bei Unsicherheiten empfehlen wir, im Vorfeld mit der FDA zu sprechen. Dafür bietet sich das Pre-Submission Meeting an. Sie können Ihre Predicate-Device-Strategie vorstellen und bereits vor Einreichung Feedback von der FDA einholen. Auf diese Weise vermeiden Sie unnötige Verzögerungen und große Unsicherheiten bei der Zulassung.
Tipp 3: Sich nicht auf eine 510(k)-Summary verlassen
Je nach Einreichung und Hersteller enthält die 510(k) Summary eines möglichen Predicate Device mal mehr und mal weniger Informationen. Manchmal findet sich z. B. eine detaillierte Vergleichstabelle zwischen dem neuen Produkt und dem Predicate Device, manchmal nur allgemeine Informationen zur Äquivalenz.
Letzteres kann einen Vergleich erschweren und das 510(k)-Verfahren gefährden. Versuchen Sie dann, über andere Wege an die notwendigen Informationen zu gelangen, z. B. über Gebrauchsanweisung, Service-Manual oder die Unternehmens-Webseite. Manchmal kann es notwendig sein, mit dem Predicate Device vergleichende Tests durchzuführen und selbst die notwendigen Vergleichsdaten zu generieren.
Tipp 4: Nichts verheimlichen
Verheimlichen Sie keine Informationen, z. B. zu kritischen Unterschieden. Wenn die FDA später davon erfährt, kann das zu ernsthaften Schwierigkeiten führen. Falls Ihnen für bestimmte Leistungsparameter keine Informationen zum Predicate Device vorliegen und Sie keine Möglichkeit sehen, diese zu generieren, dann geben Sie das so in der Einreichungsakte an.
Die FDA hat Zugriff auf die vollständige 510(k)-Akte des Predicate Device und kann bei Bedarf auf die entsprechenden Informationen zugreifen (auch wenn dies nicht der optimale Weg ist).
Tipp 5: In Sonderfällen klinische Daten einreichen
Im Regelfall benötigen Sie keine klinischen Daten für eine erfolgreiche 510(k)-Clearance. Es gibt aber Fälle, in denen klinische Daten notwendig sind.
Es gibt Unterschiede bei den Indikationen
Wenn das Predicate Device beispielsweise für eine zusätzliche Patientenpopulation mit höheren Risiken (z. B. Kinder) eingesetzt werden soll, dann können klinische Daten notwendig sein, um die Äquivalenz und somit das gleiche Maß an Sicherheit und Wirksamkeit nachzuweisen.
Für die Software zur Beurteilung von Anomalien anhand von Bilddaten heißt das: Da im neuen Produkt eine andere anatomische Region adressiert wird, sind in diesem Fall wahrscheinlich klinische Daten (echte Bilddaten) notwendig, um die Wirksamkeit, d. h. die erfolgreiche Beurteilung durch den AI-Algorithmus nachzuweisen und somit den Nachweis der Äquivalenz zu ermöglichen.
Es gibt Unterschiede in der Technologie
Unterschiedliche Materialien, insbesondere bei invasiven Produkten, können die Erhebung klinischer Daten notwendig machen, um die Sicherheit nachzuweisen.
Ein implantierbares Produkt, als Predicate Device aus nicht-resorbierbarem Material, im neuen Produkt aus Material, das im Laufe der Zeit vom Körper resorbiert wird. In diesem Fall können klinische Studien notwendig sein, um die Sicherheit am Patienten nachzuweisen.
Die Äquivalenz kann nicht allein durch nicht-klinische Tests nachgewiesen werden
Es gibt den Fall, das kein Modell für nicht-klinische Benchtests oder Tiertests zur Verfügung steht.
Ein Phantom, welches nicht verfügbar ist für eine Simulation eines bestimmten Organs für eine bestimmte Bild-Modalität. In diesem Fall sollten echte klinische Bilddaten genutzt werden.
Es gibt neu aufgetretene oder höhere Risiken des Predicate Device
Gibt es zum Predicate Device Hinweise auf initial nicht betrachtete Gefährdungen oder falsch abgeschätzte Risiken, kann dies die Generierung von klinischen Daten notwendig machen, auch wenn das Predicate Device ohne solche Daten zugelassen wurde.
Ein Produkttyp, zu dem in der Post-Market-Phase vermehrt schwerwiegende Sicherheitsprobleme gemeldet wurden. In diesem Fall ordnete die FDA PMS-Studien zu den bereits zugelassenen Produkten an und fordert die entsprechenden klinischen Nachweise zur 510(k)-Einreichung.
6. Fazit und Zusammenfassung
Die Auswahl eines geeigneten Predicate Device ist häufig anspruchsvoll und entscheidet über den Erfolg einer 510(k)-Clearance. Die FDA modernisiert dieses Verfahren und stellt strengere Anforderungen an die Auswahl eines Predicate Device. Dies erschwert den meistgenutzten Zulassungsweg in den USA.
Wir helfen Ihnen gerne bei der Auswahl geeigneter Predicate Devices und bestimmen gemeinsam mit Ihnen, ob ein 510(k)-Verfahren erfolgversprechend ist oder nicht. Wir erarbeiten und dokumentieren den Substantial-Equivalence-Vergleich. Bei Unsicherheiten organisieren und begleiten wir Sie zu einem Pre-Submission Meeting der FDA.