
Das Premarket Approval (PMA-Verfahren) müssen v. a. Hersteller von Medizinprodukten der Klasse III durchlaufen. Es ist das aufwändigste „Zulassungsverfahren“ der FDA, dessen typische Bearbeitungszeit länger als ein Jahr beträgt. Die FDA genehmigt pro Jahr meist nur wenige Dutzend PMA-Anträge.
Dieser Fachartikel stellt die Anforderungen der FDA vor und gibt Tipps, wie Hersteller diese hohe Hürde dennoch meistern können.
1. Premarket Approval (PMA): Die Grundlagen
1.1 Anwendung des PMA-Verfahrens
Die FDA setzt v. a. für Medizinprodukte der Klasse III ein PMA voraus. Das sind Produkte, die lebensrettend sind, schwerwiegende Gesundheitsbeeinträchtigungen verhindern oder ein signifikantes Gesundheitsrisiko darstellen. Beispiele sind Herzklappen, komplexe diagnostische Tests oder lebensunterstützende Geräte.
Prüfen Sie, ob die Product Classification Database der FDA bereits Ihr Produkt enthält.
1.2 Regulatorischer Rahmen
Der Titel 21 Part 814 des Code of Federal Regulations (CFR) beschreibt das PMA-Verfahren und spezifiziert die Anforderungen, die Hersteller bei diesem Verfahren erfüllen müssen.
Weitere regulatorische Anforderungen sind zu erfüllen, wie die an
- das Labeling (21 CFR Part 801),
- das Qualitätsmanagementsystem (21 CFR Part 820),
- klinische Studien (21 CFR Part 812).
1.3 Typen von PMA-Verfahren
Die FDA unterscheidet mehrere Typen von PMA-Verfahren:
| Name des PMA-Verfahrens | Beschreibung | Anwendung, Eignung |
| Original PMA | Verfahren, bei dem der Hersteller alle erforderlichen Unterlagen in einem Schritt bei der FDA einreicht | Dieser Typ wird in der Regel angewendet, wenn das Gerät bereits abschließend getestet wurde und in anderen Ländern mit bestehenden Anforderungen zugelassen ist. |
| PDP (Product Development Protocol) | Verfahren, bei dem die Entwicklung und Prüfung eines Produkts zusammengeführt werden, um die Marktzulassung zu erreichen. Der PDP ist quasi ein Vertrag zwischen einem Hersteller und der FDA. Er legt fest, welche Design- und Entwicklungsaktivitäten nötig sind, inklusive spezifischer Meilensteine und Akzeptanzkriterien. Sobald ein PDP von der FDA als vollständig anerkannt wird, gilt er als genehmigtes PMA, was die formelle Zulassung des Produkts für den Markt bedeutet. | Gut geeignet für Produkte mit etablierten und bekannten Technologien |
| Modular PMA | Bei dieser Vorgehensweise wird das Zulassungsverfahren in einzelne, genau definierte Teilbereiche (Module) gegliedert. Jedes Modul wird direkt nach seiner Fertigstellung bei der FDA eingereicht. Dadurch kann das Verfahren im Laufe der Zeit schrittweise bearbeitet werden. Das bietet den Vorteil, zeitnah Rückmeldung von der FDA zu erhalten. | Der Ansatz eignet sich insbesondere für Geräte, die sich noch in den Anfangsstadien der klinischen Studien befinden. |
Falls Sie unsicher sind, welcher Typ für Sie passend ist, dann besprechen Sie vor der Einreichung offene Punkte mit der FDA, z. B. im Rahmen eines Pre-Submission-Verfahrens. Das Johner Institut unterstützt und begleitet Sie bei diesen Pre-Submissions.
1.4 Kosten und Dauer
Für das FY 2025 betragen die Kosten für einen PMA-Antrag:
Standardgebühr: USD 540.783,00
Small-Business-Gebühr: USD 135.196,00
Die FDA zielt darauf ab, PMA-Verfahren innerhalb von 180 Tagen (Decision with no committee input) bzw. 320 Tagen (Decision with committee input) zu prüfen.
2. Inhalte eines PMA-Antrags
Ein vollständiger PMA-Antrag enthält sowohl administrative als auch technische Dokumente.
2.1 Administrative Dokumente
Der administrative Teil eines PMA-Antrags umfasst:
- Unterschriebenes Anschreiben mit Firmen- und Produktdetails
- Für internationale Hersteller: Gegenzeichnung durch den autorisierten US-Vertreter
- 10- bis 15-seitige Antragszusammenfassung mit Indikationen, Gerätebeschreibung, alternativen Behandlungsmethoden, Marketinghistorie sowie einer Übersicht der durchgeführten Studien
Zusätzlich sind erforderlich:
- Ein detailliertes Inhaltsverzeichnis
- Die finanzielle Bescheinigung oder Offenlegungen der klinischen Prüfer (soweit zutreffend)
Sorgen Sie für eine klar strukturierte Einreichung, die mit einem detaillierten Inhaltsverzeichnis versehen ist. Verwenden Sie die Checkliste der FDA zur Überprüfung der Vollständigkeit oder nutzen Sie das eSTAR-Template.
2.2 Technische Dokumentation
Die technische Dokumentation bildet das Kernstück der PMA. Die geforderten Inhalte sind in 21 CFR Part 814.20 zu finden. Sie dienen als lückenloser wissenschaftlicher Nachweis der Sicherheit und Wirksamkeit und decken mehrere Bereiche ab.
2.2.1 Fachliche Informationen
Zu den fachlichen Informationen zählen:
- Präzise Zweckbestimmung: Diese muss eine Beschreibung der Krankheit oder des Zustands, den das Produkt diagnostizieren, behandeln, verhüten, heilen oder lindern soll, sowie eine Beschreibung der Patientengruppe, für die das Produkt bestimmt ist, beinhalten.
- Vollständige Produktbeschreibung: Diese umfasst die komplette Beschreibung des Produkts und seiner funktionellen Komponenten oder Bestandteile, Verweise auf angewandte Standards und eine Umweltverträglichkeitsprüfung gemäß 21 CFR 25.20(n).
- Vollständiges Design History File (DHF)
- Risikomanagement-Dokumentation nach ISO 14971
- Kopien des Labelings, z. B. der Begleitmaterialien
- QM-Informationen zu Prozessen und Methoden der Herstellung, Verarbeitung, Verpackung, Lagerung und gegebenenfalls Installation des Produkts sowie der dafür verwendeten Einrichtungen und Kontrollen
Meistens besteht die FDA auch auf der Einreichung eines repräsentativen Produktmusters.
2.2.2 Wissenschaftliche Nachweise
Die wissenschaftlichen Nachweise umfassen:
- Präklinische Studiendaten
- Klinische Studienergebnisse
- Sicherheits- und Wirksamkeitsnachweise
- Pädiatrische Nutzungsdaten (falls relevant)
Die präklinischen und klinischen Studiendaten bilden das Herzstück der PMA. Sie müssen wissenschaftlich fundierte Nachweise zur Sicherheit und Wirksamkeit des Produkts in allen relevanten Aspekten liefern. Dies umfasst:
- Vorklinische Tests (z. B. Biokompatibilität, mechanische & physikalische Prüfungen, V&V oder Haltbarkeitsstudien)
- Klinische Erfahrungen
- Ergebnisse klinischer Studien einschließlich detailliertem Studienprotokoll, statistischer Analysen, Patientendaten, Sicherheits- und Wirksamkeitsnachweise sowie Dokumentation unerwünschter Ereignisse
- Nutzen-Risiko-Bewertung
3. Anforderungen an das QM-System
3.1 Regulatorischer Rahmen
Voraussetzung für die PMA-Einreichung ist ein Qualitätsmanagementsystem (QMS), das konform ist mit 21 CFR Part 820. Zudem muss es die Anforderungen z. B. aus Part 803 (Medical Device Reporting) und Part 806 (Reports und Corrections) erfüllen.
Begründung: Das QM-System muss sicherstellen, dass das Medizinprodukt konform mit den PMA-spezifizierten Anforderungen hergestellt wird.
3.2 Prüfung des QM-Systems
3.2.1 Einreichung von Unterlagen
Die FDA fordert ausgewählte Dokumente des QM-Systems zur Herstellung, Verarbeitung, Verpackung und Lagerung der Produkte mit dem PMA-Antrag. Zusätzlich führt sie eine detaillierte Vor-Ort-Inspektion durch:
3.2.2 Preapproval Inspection
Bereits während des Review-Verfahrens kann die FDA eine Preapproval Inspection anordnen. Diese Inspektion prüft umfassend die Einhaltung regulatorischer Anforderungen, die Prozessqualität und Dokumentationsstandards.
Besondere Bedeutung kommt während der Inspektion der Prozessvalidierung zu, da sie die konsistente Sicherheit und Qualität der Produkte gewährleistet. Zum Zeitpunkt des PMA-Antrags hat der Hersteller möglicherweise noch nicht alle erforderlichen Prozessvalidierungen abgeschlossen, sodass diese von CDRH nicht überprüft werden können. Bei der Inspektion wird erwartet, dass der Hersteller alle validierungspflichtigen Prozesse vollständig validiert hat. Sollte während der Inspektion festgestellt werden, dass dies nicht der Fall ist, wird der Inspector die Inspektion möglicherweise beenden.
3.2.3 Postapproval Inspection
Die Postapproval Inspection erfolgt etwa 8 bis 12 Monate nach Marktzulassung und kontrolliert die kontinuierliche Einhaltung von Qualitätsstandards und Änderungsprozessen sowie die korrekte Umsetzung ursprünglich genehmigter Herstellungs- und Entwicklungsspezifikationen.
4. Ablauf des PMA-Verfahrens
4.1 Einreichung bei der FDA
Die PMA-Einreichung erfolgt elektronisch über das CDRH-Portal an die FDA und wird entweder im eSTAR-Format (elektronisches, strukturiertes Antragsformat mit vordefinierten Feldern) oder als eCopy (elektronische Kopie Ihres Antrags) gesendet.
Die Erstellung im eSTAR-Format ist für PMA-Anträge freiwillig und auch nur möglich für Original PMA. PDPs und Modular PMAs müssen über das eCopy-Verfahren eingereicht werden.
4.2 Das Review-Verfahren
Ein PMA wird durch ein interdisziplinäres Team geprüft, das aus 10 bis 15 Mitgliedern besteht. Geprüft wird in einem mehrstufigen Prozess, der drei Ebenen (regulatorisch, wissenschaftlich, Qualitätsmanagement) umfasst.
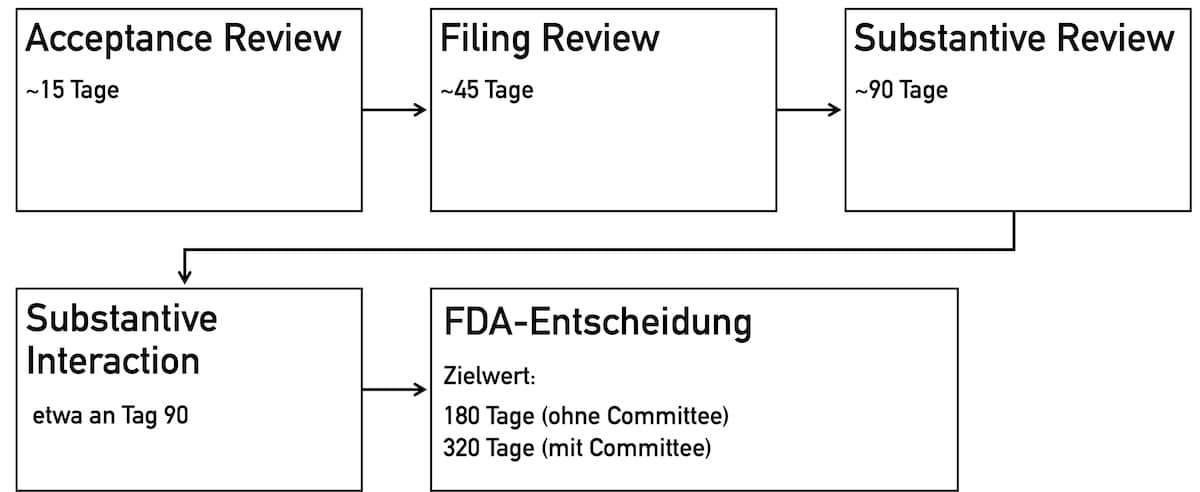
4.2.1 Acceptance Review
Beim Acceptance Review handelt es sich um einen administrativen Prozess. Es wird geprüft, ob alle notwendigen Unterlagen vorhanden sind. Der Antragsteller erhält innerhalb von 15 Tagen per E-Mail eine Bestätigung, ob der Antrag angenommen ist oder nicht. Im letzten Fall erhält der Antragsteller eine Abweichungsliste (entfällt beim eSTAR-Verfahren).
4.2.2 Filing Review
Das Filing Review stellt eine Prüfung der Inhalte dar, um festzulegen, ob der Antrag ausreichend vollständig ist, um weiter bearbeitet zu werden. Innerhalb von 45 Tagen erhält der Antragsteller eine Benachrichtigung per E-Mail, ob die Unterlagen für eine detaillierte fachliche Prüfung akzeptiert sind bzw. welche Informationen fehlen.
4.2.3 Substantive Review
Im Substantive Review findet eine eingehende Prüfung der wissenschaftlichen und regulatorischen Aspekte sowie des Qualitätsmanagementsystems statt. Während des Prüfprozesses arbeitet die FDA eng mit dem Antragsteller zusammen, um identifizierte Mängel gezielt und fristgerecht zu beheben.
Stellen Sie sicher, dass Sie ausreichend Ressourcen haben, um Rückfragen der FDA zeitnah und ausführlich beantworten zu können.
4.2.4 Substantive Interaction
Während der Phase Substantive Interaction gibt die FDA dem Antragsteller Feedback und entscheidet über den Verlauf der weiteren Bearbeitung:
- Weiterbearbeitung der PMA im interaktiven Review mit dem Antragsteller ODER
- Ausstellung eines Major Deficiency Letters und Zurückstellen der PMA, bis die Abweichungen behoben sind
Sie haben zu diesem Zeitpunkt die Möglichkeit, ein Day-100 Meeting durchzuführen. Weiterhin ist es möglich, ein Q-Submission Request einzureichen, um den Fortschritt der Einreichung oder aufgetretene Abweichungen zu besprechen.
Bereits in dem Antrag für die PMA kann ein Day-100 Meeting mit der FDA angefragt werden. Beantragen Sie dieses immer gleich mit! Sollten Sie es nicht benötigen, können Sie absagen. Wird es doch erforderlich, sparen Sie wertvolle Zeit!
4.2.5 Approval-Entscheidung
Die Entscheidung für oder gegen ein Approval trifft die FDA in der Regel nach 180 Tagen bzw. 320 Tagen. Folgende Möglichkeiten gibt es:
- Approval Order: Produkt kann verkauft werden
- Approval Pending Deficiencies Letter: Produkt kann nicht verkauft werden. Die Entscheidung adressiert Abweichungen, die vor Freigabe der PMA behoben werden müssen.
- Approval Pending GMP Letter: Produkt kann nicht verkauft werden. Das QM-System ist noch nicht in Übereinstimmung mit den FDA-Anforderungen.
- Not-approvabale Letter: Das Produkt kann nicht verkauft werden. Es gibt größere Mängel, die neue klinische oder präklinische Daten erfordern können.
Die Prüfdauer für die PMA-Einreichung ist davon abhängig, ob ein Advisory Committee einbezogen werden muss oder nicht. Ohne Einbeziehung eines Komitees arbeitet die FDA darauf hin, PMAs innerhalb von 180 Tagen zu prüfen. Mit Einbeziehung des Advisory Committees verlängert sich die geplante Zeit bis zur Entscheidung auf 320 Tage.
5. Postapproval-Anforderungen
Nach der Zulassung kann die FDA dem Hersteller weiterführende Anforderungen auferlegen, deren Erfüllung für den Marktzugang entscheidend ist.
5.1 Einschränkungen
Diese Anforderungen können Einschränkungen bei Vertrieb und Nutzung, verpflichtende Sicherheitsberichte sowie spezifische Kennzeichnungs- und Dokumentationspflichten umfassen.
5.2 Studien
Für bestimmte Produkttypen, z. B. pädiatrische Produkte, kann die FDA zusätzliche Studien zur Postmarket Surveillance (PMS-Studien) anordnen.
5.3 Berichte
Jährliche Berichte sind ein zentrales Element der Postapproval-Überwachung und müssen Veränderungen, wissenschaftliche Publikationen und neue Erkenntnisse zur Sicherheit und Wirksamkeit des Medizinprodukts dokumentieren. Der Bericht ermöglicht der FDA eine umfassende Risikobewertung.
5.4 Produktänderungen
Wesentliche Produktänderungen, die die Sicherheit oder Wirksamkeit beeinflussen könnten, erfordern die Einreichung eines PMA Supplements zur FDA-Prüfung. Dies gilt insbesondere bei unerwarteten Nebenwirkungen, Geräteausfällen oder geplanten Änderungen an Herstellung, Konstruktion oder Kennzeichnung des Medizinprodukts.
Die FDA unterscheidet verschiedene Supplement-Typen, welche sich nach der Signifikanz der Änderung und deren Auswirkung auf die Sicherheit des Produkts richten:

| PMA-Supplement-Typ | Erklärung |
| Panel-Track PMA Supplement | Eine Änderungsmitteilung zu einem genehmigten Original-PMA- oder Premarket-Report, die eine wesentliche Änderung der Auslegung oder Leistung des Produkts oder eine neue Indikation darstellt und für die klinische Daten erforderlich sind, um die Sicherheit und Wirksamkeit nachzuweisen. |
| 180-Day PMA Supplement | Eine Änderungsmitteilung zu einem genehmigten Original-PMA- oder Premarket-Report, bei der es sich nicht um eine Panel-Track-Ergänzung handelt und die eine wesentliche Änderung der Komponenten, Materialien, des Designs, der Spezifikationen, der Software, der Farbzusätze oder der Kennzeichnung darstellt. |
| Real-Time PMA Supplement | Eine Änderungsmitteilung zu einem genehmigten Original-PMA- oder Premarket-Report, welche eine geringfügige Änderung in der Auslegung des Produkts, der Software, der Sterilisation oder im Labeling darstellt. Der Antragsteller hat eine genehmigte Besprechung bei der FDA beantragt, um das Supplement gemeinsam mit der FDA zu prüfen. |
| Special PMA Supplement | Eine Änderungsmitteilung zu einem genehmigten Original-PMA- oder Premarket-Report zur Verbesserung der Sicherheit des Produkts oder der sichereren Nutzung des Produkts sowie für Labeling-Anpassungen, um die Produktsicherheit zu erhöhen. Diese Änderungen dürfen vom Antragsteller durchgeführt werden, bevor die FDA das Supplement genehmigt hat. |
| 30-Day Notice | Für Änderungen am Herstellprozess oder der Herstellmethode, welche die Sicherheit und Wirksamkeit des Produkts beeinflussen könnten |
6. Fazit und Zusammenfassung
Das PMA-Verfahren der FDA stellt für Medizinproduktehersteller eine komplexe, aber überwindbare Hürde dar.
Kernelemente sind eine lückenlose wissenschaftliche Dokumentation, umfassende präklinische und klinische Studiendaten sowie ein robustes Qualitätsmanagementsystem. Die FDA prüft nicht nur die technischen Unterlagen, sondern bewertet die Sicherheit und Wirksamkeit des Medizinprodukts in allen Aspekten.
Die Zulassung ist dabei kein einmaliger Vorgang, sondern der Beginn einer kontinuierlichen regulatorischen Verantwortung. Post-Approval-Anforderungen wie jährliche Berichte, Sicherheitsmeldungen und Änderungsmitteilungen gehören ebenso zum Prozess wie FDA-Inspektionen.
Für Unternehmen bedeutet dies: Erfolgreich sind jene, die das PMA-Verfahren nicht als administrative Hürde, sondern als strategischen Prozess zur Markteinführung verstehen.
Melden Sie sich gerne, wenn Sie Fragen rund um das Thema PMA haben. Wünschen Sie Unterstützung oder einen Austausch zu Ihrer strategischen Herangehensweise, Antworten auf Fragen zu Ihren Dokumenten oder zu administrativen Themen? Nutzen Sie unser Kontaktformular. Das Team vom Johner Institut steht Ihnen gerne zur Seite!



Herzlichen Dank für den Übersichtsartikel. Könnten Sie bitte die Kostenstruktur und die Zeitpunkte der Zahlungen etwas erläutern? Die 0,5M USD für ein PMA sind ja doch etwas unterschiedlich im Vergleich zu den 510(k) Einreichungen und müssen in dieser Höhe gewöhnlich auch in der Kostenplanung eines Entwicklungsprojektes berücksichtigt werden.
Lieber Herr Abbe,
vielen Dank für Ihre Rückfrage.
Die Kosten für den PMA werden zum Zeitpunkt der Einreichung fällig. Dies gilt auch für Modulare PMAs, bei denen mit Einreichung des ersten Moduls die User Fees fällig werden. Weitere Informationen hierzu erhalten Sie auch im FDA Guidance Document „User Fees and Refunds for Premarket Approval Applications and Device Biologics License Applications„.
Wenn Sie ein Small Business sind, müssen Sie die Genehmigung für die reduzierte Gebühr vor der Einreichung Ihres PMA erhalten. Die Prüfung der Small Business Determination durch die FDA dauert etwa 2 Monate.
Herzliche Grüße,
Margret Seidenfaden