
Den Begriff „Probenahme-Set“ definiert weder die IVDR noch eine andere Regularie. Dennoch gibt es (indirekte) regulatorische Anforderungen, die IVD-Hersteller und medizinische Labore kennen und beachten müssen. Die Anforderungen hängen von der jeweiligen Konstellation ab.
Dieser Artikel stellt fünf Konstellationen vor. Er verschafft damit Klarheit und hilft, regulatorischen Ärger zu vermeiden und regulatorische Aufwände zu minimieren.
1. Probenahme-Set: Definition & Beispiele
a) Definition
In Ermangelung einer offiziellen Definition legen wir für diesen Artikel fest:
Ein Probenahme-Set ist eine Zusammenstellung von Produkten, welche laut Inverkehrbringer gemeinsam von einem Anwender dazu verwendet werden, um menschliche Proben (z. B. Blut, Speichel, Gewebe, Urin) entnehmen und für weitere in-vitro-diagnostische Untersuchungen fachgerecht lagern und/oder transportieren zu können.
Definition: Johner Institut
Mit der Revision 3 des Dokuments MDCG 2020-16 hat die Medical Device Coordination Group ‚Kit‘ definiert:
Kit means a set of components that are packaged together and intended to be used to perform a specific in vitro diagnostic examination, or a part thereof.
Definition: MDCG 2020-16 rev.3
Diese Definition ist auf Probenahme-Sets ebenso anwendbar, da sie einen Teil einer in-vitro-diagnostischen Untersuchung darstellen. Die Anwendbarkeit geht auch aus dem überarbeiteten Text zur Regel 5 (c) in MDCG 2020-16 rev.3 hervor.
b) Beispiel
Probenahme-Sets für eine Gewebeprobe können beispielsweise die folgenden Produkte umfassen:
- Biopsiestanze
- Desinfektions-Pad
- Probengefäß mit Fixationslösung
- Transportverpackung
- Aufkleber mit Patienten-ID und Empfänger-Adresse
- Gebrauchsanweisung
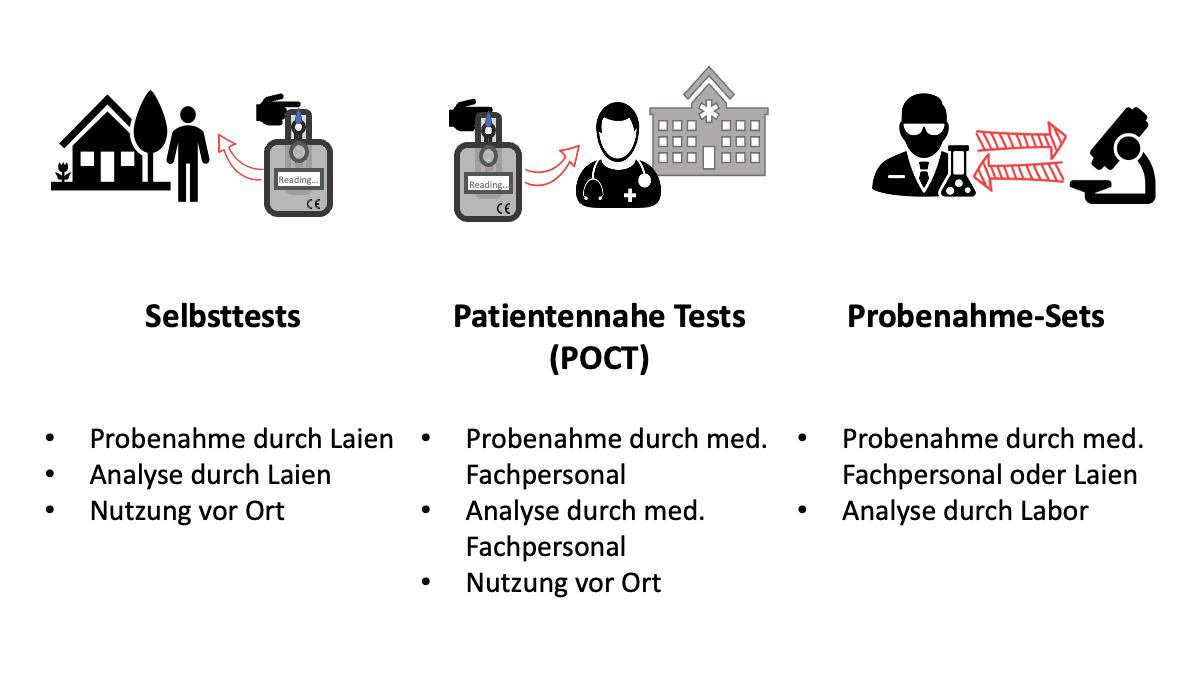
c) Abgrenzung Probenahme-Sets, patientennahe Tests und Selbsttests
Probenahme-Sets können sowohl bei der Probenentnahme zuhause als auch in einer Gesundheitseinrichtung zur Anwendung kommen, z. B. in einer Arztpraxis.
Dabei kann die Probe – je nach Zweckbestimmung – durch den Patienten selbst entnommen werden (wie bei einem Produkt zur Eigenanwendung, auch Selbsttest genannt) oder durch eine medizinische Fachkraft (wie bei einem Produkt für patientennahe Tests).
Was die Probenahme-Sets von den Selbsttests und den patientennahen Tests unterscheidet, ist der Zeitpunkt der Diagnostik. Sie erfolgt erst nach Versand der Probe in ein medizinisches Labor und wird dort von Laborpersonal durchgeführt und nicht an Ort und Stelle der Probenahme.
Beispiel: Tests auf eine Infektion mit SARS-CoV-2
Probenahme-Set: Von einem medizinischen Labor an einen niedergelassenen Arzt bzw. einem Angehörigen der Gesundheitsberufe bereitgestelltes Set mit Rücksendebox zum Versand der professionell entnommenen Probe an das Labor zur Durchführung eines PCR-Tests auf SARS-CoV-2 RNA
Probenahme-Set (DTC): Im stationären Handel oder online gekauftes Probenahme-Set mit Rücksendebox zum Versand einer durch den Laien selbst entnommenen Probe an ein Labor zur Durchführung eines PCR-Tests auf SARS-CoV-2 RNA
Patientennaher Test (POCT): Antigen-Schnelltest in einem sog. Corona-Testzentrum
Selbsttest: Antigen-Test zur Eigenanwendung mit eigener Probenahme, gekauft z. B. im Supermarkt, in der Apotheke oder online
d) Kontext, in dem Probeentnahme-Sets zum Einsatz kommen
Inverkehrbringung durch Gesundheitseinrichtungen (Labore)
Probenahme-Sets werden oft nicht von klassischen IVD-Herstellern in Verkehr gebracht, sondern von Gesundheitseinrichtungen wie medizinischen Laboratorien, die in-vitro-diagnostische Untersuchungen anbieten und somit selbst Betreiber von IVD sind.
Diese Labore versenden die Probenahme-Sets im Normalfall an Arztpraxen. Die Ärzte entnehmen dann mit den im Set zur Verfügung gestellten Produkten die Probe und senden sie im ebenfalls im Set befindlichen Probenbehältnis an das Labor zurück.
Diese Praxis wird vereinzelt auch von CE-IVD Herstellern mit angeschlossenem medizinischem Labor angewendet.
Direct-to-Consumer Testing (DTC-Tests)
Eine Besonderheit, die meist bei genetischen Tests zu finden ist, ist der direct-to-consumer-Test (DTC-Test). Bei dieser Variante wird das Probenahme-Set nicht an eine Arztpraxis oder an eine andere Gesundheitseinrichtung versandt, sondern direkt an den Patienten: Dieser bestellt das Set im Internet oder erwirbt es im Einzelhandel.
Probenahme, Verpackung und Rückversand erfolgen hier durch den Laien. Falls der Laie weitere Tätigkeiten nach der Probenahme durchführen muss, kann die Schwelle zum Selbsttest überschritten werden, was weitreichende regulatorische Folgen nach sich zieht.
DTC Testing kann sowohl von medizinischen Laboren (reine Betreiber oder Betreiber mit Inhouse-IVD) als auch von Herstellern von CE-IVD mit angeschlossenem medizinischem Labor angeboten werden.
Die FDA definiert DTC-Tests folgendermaßen:
In vitro diagnostics (IVDs) that are marketed directly to consumers without the involvement of a health care provider are called direct-to-consumer tests (also referred to as DTC). These tests generally request the consumer collect a specimen, such as saliva or urine, and send it to the company for testing and analysis.
2. Regulatorische Einordnung von Probenahme-Sets
Probenahme-Sets führen keine Diagnostik durch – im Gegensatz zu den Selbsttests und den patientennahen Tests. Daher werden sie regulatorisch anders als die anderen beiden Produkttypen gehandhabt.
Die Anforderungen an ein Probenahme-Set hängen von dessen regulatorischer Einordnung und diese wiederum von den Inhalten der Sets ab.

Die regulatorische Einordnung ist nicht trivial, weil weder die IVDR noch andere Regularien den Begriff „Probenahme-Set“ definieren.
Einrichtungen, die Probenahme-Sets an Angehörige der Gesundheitsberufe („Healthcare Professionals“) in z. B. Arztpraxen oder direkt an Patienten versenden oder an den Einzelhandel ausliefern, müssen beurteilen, welche Art von Produkt sie in den Verkehr bringen. Handelt es sich um eine dieser fünf Konstellationen?
- Behandlungseinheit
- Medizinprodukt bzw. ein In-vitro-Diagnostikum
- Zubehör
- DTC-Test
- vereinzeltes Produkt
Die folgenden Abschnitte beschreiben diese Konstellationen.
a) Probenahme-Set als Behandlungseinheit
Definition
Eine Behandlungseinheit besteht, wenn das Probenahme-Set eine Zusammenstellung von einem oder mehreren CE-gekennzeichneten Medizinprodukten und IVDs oder Nicht-Medizinprodukten ist.
In dieser Kombination stellt ein Probenahme-Set in den allermeisten Fällen eine Behandlungseinheit im Sinne der MDR dar.
„Behandlungseinheit“ bezeichnet eine Kombination von zusammen verpackten und in Verkehr gebrachten Produkten, die zur Verwendung für einen spezifischen medizinischen Zweck bestimmt sind;
MDR Art. 2 (10)
Die IVDR kennt das Konzept der Behandlungseinheit nicht. Zwar zählen IVDs als Medizinprodukte:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das…“
IVDR, Artikel 2 (2)
Allerdings fühlt sich die MDR nicht für IVDs zuständig:
„Diese Verordnung gilt nicht für a) In-vitro-Diagnostika im Sinne der Verordnung (EU) 2017/746.“
MDR, Artikel 1(6)
Dennoch lässt sich das Konzept der „Behandlungseinheit“ auch auf IVDs übertragen, sofern das Set eben mindestens ein nach MDR (oder MDD) CE-gekennzeichnetes Medizinprodukt enthält, zumal die Definition auf Probenahme-Sets sehr gut passt und die MDR explizit auch IVDs als Bestandteile von Behandlungseinheiten vorsieht (s. Artikel 22 MDR, Abschnitt 1).
Falls das Probenahme-Set auch ein Medizinprodukt im Sinne der MDR enthält, ist das Konzept der Behandlungseinheit unter den unten genannten Voraussetzungen anwendbar.
Ein Beispiel ist ein Set zur Blutabnahme, bestehend aus einem Medizinprodukt, dem Butterfly (Flügelkanüle mit Schlauch und Adapter) und einem Probenbehältnis als IVD (Blutröhrchen).
Enthält das Probenahme-Set kein CE-markiertes Medizinprodukt, sondern z. B. ausschließlich IVDs, ist eine Inverkehrbringung nach MDR Art. 22 nicht möglich.
Voraussetzungen
Die Voraussetzungen dafür, dass ein Probenahme-Set als Behandlungseinheit und nicht als Medizinprodukt bzw. IVD zählt, sind:
- Mindestens eines der Produkte im Set ist ein Medizinprodukt oder ein Zubehör dazu.
- Alle Produkte im Set werden im Rahmen ihrer ursprünglichen Zweckbestimmung eingesetzt.
- Alle Produkte sind konform mit den für sie geltenden regulatorischen Anforderungen und tragen beispielsweise eine CE-Kennzeichnung.
Für genetische Tests verschicken die Hersteller sterile Abstrichtupfer („Wattestäbchen“), die als Medizinprodukt qualifiziert sind, sowie ein mit Puffer gefülltes Transportröhrchen, das als Probenbehältnis und damit als IVD zählt. Diese Kombination ist eine Behandlungseinheit im Sinne der MDR.
Regulatorische Anforderungen an Behandlungseinheiten
Inverkehrbringer von Behandlungseinheiten müssen dafür keine Konformitätsbewertung durchlaufen und auch keine CE-Kennzeichnung an die Behandlungseinheit anbringen. Aber sie müssen die Pflichten erfüllen, die Artikel 22 der MDR ihnen auferlegt zuzüglich UDI und Registrierung.
Gegebenenfalls unterliegen die Inverkehrbringer den Pflichten von Händlern. Dann müssen sie Artikel 14 und ggf. Artikel 16 der MDR beachten.
Lesen Sie hier mehr zu den Pflichten der Händler.
b) Probenahme-Set als IVD
Die IVDR legt fest, dass Probenbehältnisse als IVD gelten:
„Probenbehältnisse gelten als auch In-vitro-Diagnostika;“
IVDR, Artikel 2(2)
Die Verordnung definiert auch den Begriff Probenbehältnis:
bezeichnet ein luftleeres wie auch sonstiges Produkt, das von seinem Hersteller speziell dafür gefertigt wird, aus dem menschlichen Körper stammende Proben unmittelbar nach ihrer Entnahme aufzunehmen und im Hinblick auf eine In-vitro-Untersuchung aufzubewahren;
(Falls Ihnen diese Definition unklar erscheint, dann lesen Sie die englische Version.)
Ein Probenahme-Set kann auch aus einer Kombination von zwei IVDs bestehen, z. B. einem Probenröhrchen und einem Puffer. Wird dieses Probenahme-Set nun von einem Labor an seine Kunden (Angehörige der Gesundheitsberufe) verschickt, dann liegt eine Inverkehrbringung eines IVDs – zumindest der Klasse A – vor. In solchen Fällen muss das Labor die IVDR vollumfänglich anwenden.
Der Fachartikel zur IVDR stellt deren Anforderungen vor. Ein weiterer erläutert die Klassifizierung von IVDs.
Allerdings enthält ein Probenahme-Set typischerweise nicht nur ein oder mehrere Probenbehältnisse (und damit ein oder mehrere IVDs), sondern weitere Produkte. Dann besteht die Möglichkeit, dass dieses Probenahme-Set selbst nicht als IVD zu betrachten ist. Wir empfehlen Ihnen, dies bei Ihren Probenahme-Sets zu prüfen, um die korrekte Umsetzung der regulatorischen Anforderungen sicherzustellen.
c) Probenahme-Set als Zubehör
Auch eine Qualifizierung von Probenahme-Sets als Zubehör steht im Raum.
Ein Gegenstand, der zwar an sich kein In-vitro-Diagnostikum ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten In-vitro-Diagnostika verwendet zu werden, und der speziell dessen/deren Verwendung gemäß seiner/ihrer Zweckbestimmung(en) ermöglicht oder mit dem die medizinische Funktion des In-vitro-Diagnostikums/der In-vitro-Diagnostika im Hinblick auf dessen/deren Zweckbestimmung(en) gezielt und unmittelbar unterstützt werden soll;
IVDR, Artikel 2(4)
Diese Qualifizierung wäre nur dann anzuwenden, wenn das Set spezielle IVDs gezielt unterstützen soll und selbst keine IVDs enthält. Meist ist eine solche Qualifizierung als Zubehör nicht möglich oder sinnvoll, weil
- Probenbehältnisse IVDs sind,
- das Set selten eine Zweckbestimmung hat, die gezielt die Zweckbestimmung konkreter IVDs unterstützen soll,
- diese Qualifizierung mit dem Ansatz der Behandlungseinheiten gemäß Artikel 22 der MDR vermieden werden kann.
Zudem wird Zubehör ohne kritische Merkmale gemäß Regel 5a) den IVDs der Klasse A zugeordnet, womit die IVDR entsprechend umfänglich einzuhalten ist.
d) Probenahme-Sets für DTC-Tests
Bei einem Direct-to-Consumer-Test werden die Probenahme-Sets direkt an den Patienten gesendet, damit diese (als Laien) die Proben entnehmen.
Die Inverkehrbringung solcher Sets als Behandlungseinheit nach Artikel 22 MDR ist nur möglich, sofern die darin befindlichen Produkte (mit Ausnahme des Probenbehältnisses) in ihrem jeweiligen bestimmungsgemäßen Gebrauch auch für die Verwendung durch Laien vorgesehen sind.
Daher sollte der Inverkehrbringer bei der Zusammenstellung der Probenahme-Sets und der Benutzerhinweise besondere Sorgfalt walten lassen und auch Gebrauchstauglichkeitsstudien in Betracht ziehen.
War eine Verwendung der Bestandteile des Sets (bis auf das Probenbehältnis, siehe Manual on Borderline and Classification in the Community Regulatory Framework for Medical Devices Version 1.22 05-2019, Kapitel 2.1 letzter Absatz) durch Laien nicht vorgesehen, muss das Probenahme-Set als CE-markiertes Produkt zur Eigenanwendung in Verkehr gebracht werden. Somit würde das Set gemäß IVDR, Anhang VIII, Regel 4 mindestens in Klasse C fallen.
Dieser Fachartikel zu den Selbsttests stellt die regulatorischen Anforderungen daran vor. Fluch und Segen von DTC diskutiert auch das Ärzteblatt.
Erfüllen alle weiteren Bestandteile des Sets die Anforderungen an eine Eigenanwendung durch Laien, kann das Set dennoch nur als Behandlungseinheit nach MDR Artikel 22 oder als IVD einer Klasse niedriger als Klasse C in Verkehr gebracht werden, wenn der Laie nach der Probenahme keine Tätigkeiten durchführen muss, die die Schwelle zum Selbsttest überschreiten.
Übersteigen die Tätigkeiten das Sammeln und Verpacken sowie Tätigkeiten, um die Integrität und Stabilität der Proben zu gewährleisten, wird das Set ebenso als Produkt zur Eigenanwendung klassifiziert (MDCG 2020-16 rev.3 Rule 4 (a) und Rule 5 (c); Borderline Manual Version 1.22 05-2019, Kapitel 2.1).
Beachten Sie bei genetischen Tests weitere regulatorische Vorgaben wie das Gendiagnostikgesetz (GenDG), das bestimmte diagnostische Untersuchungen nur nach vorangehender ärztlicher Beratung erlaubt.
e) Verzicht auf ein Probenahme-Set
Organisationen wie Labore können nicht nur durch den Ansatz einer „Behandlungseinheit“ strengere regulatorische Anforderungen, wie sie z. B. bei der Inverkehrbringung eines neuen IVDs zu erfüllen sind, vermeiden. So können sie erwägen, auf die Konfektionierung von Probenahme-Sets ganz zu verzichten und dennoch ihren Kunden Produkte zur Verfügung zu stellen.
Labore sollten ihre Angebote (Webseite, Produktkataloge) und Rechnungen sehr sorgfältig formulieren. Nur so stellen sie klar, ob sie ganze Sets in Verkehr bringen oder auf dem Markt nur einzelne, bereits CE-markierte Produkte bereitstellen, die aber gegebenenfalls gemeinsam ausgeliefert werden.
Falls die Labore Produkte weiterverkaufen (oder auch unentgeltlich weitergeben), müssen sie ihren Pflichten als Händler (u. a. ist das sogenannte Händler-QMS nötig) gerecht werden; diese beschreibt die IVDR in den Artikeln 14, 16, 22 und 24 (1) c).
3. Regulatorische Anforderungen
Abhängig von der regulatorischen Einordnung eines Probenahme-Sets müssen die Inverkehrbringer verschiedene regulatorische Anforderungen erfüllen. Dieses Kapitel fasst einige zusammen.
a) Anforderungen der MDR und IVDR
| Situation | Regulatorische Anforderungen |
| Inverkehrbringung von Behandlungseinheiten | MDR, Artikel 22: Systeme und Behandlungseinheiten MDR, Artikel 29(2): Registrierung von Produkten |
| Inverkehrbringen eines IVD | IVDR, Artikel 10: Allgemeine Pflichten der Hersteller |
| Bereitstellen eines CE-markierten Produkts auf dem Markt | IVDR/MDR, Artikel 14: Allgemeine Pflichten der Händler IVDR/MDR, Artikel 16: Fälle, in denen die Pflichten des Herstellers auch für Importeure, Händler oder andere Personen gelten IVDR, Artikel 22/MDR, Artikel 25: Identifizierung innerhalb der Lieferkette |
b) Leitlinien und Normen
- MDCG 2021-26 Q&A on repackaging & relabelling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746
- MDCG 2021-27 Questions and Answers on Articles 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746
- MDCG 2018-6 Clarifications of UDI related responsibilities in relation to article 16
- MDCG 2018-3 Rev.1 Guidance on UDI for systems and procedure packs
- MDCG 2018-4 Definitions/descriptions and formats of the UDI core elements for systems or procedure packs
- ISO/TS 20658:2023: Medizinische Laboratorien – Anforderungen an Probennahme, Transport, Erhalt und Handhabung von Proben
- ASTM D4169-22: Standard Practice for Performance Testing of Shipping Containers and Systems
c) Weitere Vorgaben mit Bezug zur Verpackung und zum Transport
Wird in dem Probenahme-Set auch Material zum Proben-Rückversand an das Labor bereitgestellt, müssen verschiedene internationale Vorgaben zum Versand biologischen Materials beachtet werden. Diese sind davon abhängig, welches Material (potentiell human-pathogen, infektiös etc.) von wo nach wo (Deutschland, Europa, USA etc.) und wie (Flugzeug, über Wasser/Land) versendet wird. Relevant können u. a. folgende Vorgaben sein:
- Übereinkommen über die internationale Beförderung gefährlicher Güter auf der Straße: ADR der UNECE, Artikel bei Wikipedia
- Vorgaben des Bundesverkehrsministeriums
- Vorgaben zu der Verpackungsanweisung P650
- Patientenproben richtig versenden der Berufsgenossenschaft für Gesundheitsdienst und Wohlfahrtspflege (BGW)
Sollten Labore das Probenahme-Set mit einem (eigenentwickelten) Inhouse-Verfahren kombinieren, müssen bei der Leistungsbewertung des Inhouse-IVD (IH-IVD) die Probenahme und der Transport unbedingt berücksichtigt werden. Dies schreibt die IVDR in Anhang I, Abschnitt 9.1. vor.
d) Weiterführende Informationen
Die folgenden Fachartikel können als weiterführende Literatur und bei bestimmten Fragestellungen hilfreich sein:
- Fachartikel zu den Händlerpflichten
- Fachartikel zu den Inhouse-IVD (IH-IVD, auch LDT)
- Fachartikel zur Leistungsbewertung von IVD
- Artikel des BVMed zur Vereinzelung von Medizinprodukten nach der MDR
4. Fazit und Zusammenfassung
a) Die regulatorische Einzelfallbewertung ist notwendig
Das Konzept eines „Probenahme-Sets“ kennen die Gesetze wie die IVDR nicht. Daher müssen Inverkehrbringer klären, wie diese Sets im konkreten Kontext regulatorisch einzuordnen sind. Nur so können sie die anwendbaren regulatorischen Anforderungen gezielt ermitteln und umsetzen.
b) Die regulatorischen Anforderungen unterscheiden sich stark
Je nach Einordnung sind diese Anforderungen leicht zu erfüllen (z. B. wenn nur die Händlerpflichten gelten) oder sehr hoch (z. B. wenn ein erneutes Konformitätsbewertungsverfahren durchlaufen werden muss).
c) Die Einordnung lässt sich beeinflussen
Wer verhindern will, als Inverkehrbringer eines IVDs die komplette IVDR einhalten zu müssen, konfektioniert sein Probenahme-Set so, dass die Anforderungen an Behandlungseinheiten gemäß MDR, Artikel 22 erfüllt werden. Das spart viel Aufwand.
Dieses Vorgehen setzt allerdings voraus, dass die Benutzerhinweise für das Probenahme-Set auf keinen Fall die jeweilige Zweckbestimmung der einzelnen Inhalte des Sets verändern.
Wem auch diese Anforderungen zu hoch erscheinen, sollte prüfen, ob die Inverkehrbringung eines Sets überhaupt erforderlich ist. Falls nicht, können sich die Inverkehrbringer auf eine Rolle als Händler beschränken und anstelle von neu konfektionierten Sets nur Einzelprodukte an professionelle Anwender versenden.
d) Sonderfall DTC beachten
Das ist bei Direct-to-Consumer-Tests nicht möglich. Auch hier ist es eher unwahrscheinlich, dass ein Set ausschließlich Produkte zur Eigenanwendung enthält. Dann droht eine Inverkehrbringung eines IVD der Klasse C oder sogar D.
Ist das der Fall, sind umfangreiche regulatorische Anforderungen zu erfüllen: Von einer kompletten Technischen Dokumentation gemäß Anhang II und III der IVDR inklusive einer ausführlichen Dokumentation zur Gebrauchstauglichkeit bis zur Einbeziehung einer Benannten Stelle für die Bewertung der Technischen Dokumentation und des QM-Systems beim Konformitätsbewertungsverfahren.
Die IVD-Expertinnen und -Experten des Johner Instituts helfen bei der Entwicklung einer regulatorischen Strategie, mit der Labore und andere Organisationen ihre Produkte rechtskonform in den Verkehr bringen und dabei unnötige Risiken und Aufwände (z. B. für eine Konformitätsbewertung) vermeiden können.
Melden Sie sich für eine kostenlose Abstimmung. Danach wissen Sie, welche Optionen Ihnen offenstehen.
Änderungshistorie:
- 2024-07-16:
- 1. a) Definition von ‚Kit‘ und Verweis auf MDCG 2020-16 rev.3 hinzugefügt
- 1. c) Hinzufügen einer neuen Abblidung 1
- 2. d) Verweis auf MDCG 2020-16 rev.3 hinzugefügt



Sehr geehrter Herr Hafen,
vielen Dank für diese gute Übersicht.
Wenn ich mich für die Variante „Behandlungseinheit“ entscheide, wie muss mich dann im DMDIS anmelden?
Zu meinem Erstaunen habe ich gesehen, dass es auch in 2023 noch üblich ist, sich nach §25 MPG als „Zusammensteller von Systemen und Behandlungseinheiten“ anzuzeigen. Ihrer Darstellung habe ich aber entnommen, dass ich das gar nicht machen muss nach §22. Wie verhalte ich mich richtig?
Außerdem frage ich mich, inwieweit indirekt auch für mich als Set-Kombinierer von MP und IVD innerhalb der Zweckbestimmung eine Pflicht zur ISO 13485-Zertifizierung besteht oder es wie für Händler freiwillig ist.
Zuletzt gibt es im Internet die Ansicht, dass für das Probenahme-Set eine eigene Basic-UDI DI zu vergeben sei, die dann auch registriert werden muss, und einer UDI-DI, auch wenn ich CE-gekennzeichnete Produkte innerhalb ihrer Zweckbestimmung zusammenstelle.
Sie verweisen auf Art. 16 MDR und MDCG 2018-6. Da liest es sich für mich anders.
Was ist richtig?
Vielen Dank!
Liebe Frau Müller,
herzlichen Dank für Ihre wichtigen Fragen.
MDR Artikel 29(2) gibt die Vergabe der UDI und die Registrierung in der EUDAMED für Behandlungseinheiten vor. Ich habe diese Information im Artikel ergänzt. Derzeit, da die EUDAMED noch nicht voll funktionstüchtig ist, müssen Behandlungseinheiten weiterhin im DMIDS registriert werden. Die Herleitung ist etwas komplizierter. Hierfür werden MPDG § 96 Absatz 1, MPG § 25 Absatz 1, 4 und 5 und § 33 sowie MDR, Artikel 123 Absatz 3 und die Bekanntmachung „BAnz AT 28.05.2021 B6“ im Bundesanzeiger benötigt. Das Ergebnis lautet: Behandlungseinheiten werden bis 2 Jahre nach der Bekanntmachung der vollen Funktionstüchtigkeit von EUDAMED (Art. 123 (3) e) 18 Monate nach 6 Monate nach Bekanntmachung) im DMIDS registriert und spätestens ab dann in der EUDAMED.
Als Inverkehrbringer einer Behandlungseinheit benötigen Sie kein vollständiges QM-System nach ISO 13485 und erst recht kein zertifiziertes QMS. Inverkehrbringer von Behandlungseinheiten sind aber auch Händler, da sie die in der Bahandlungseinheit kombinierten Produkte erwerben und weitergeben, und benötigen somit das sogenannte Händler-QMS. Lesen Sie mehr dazu hier unter 2. f).
Sehr geehrter Herr Hafen,
ich tue mich noch mit dem Kapitel „2e) Probenahme-Sets für DTC-Tests“ sehr schwer.
1. Der Artikel spezifiert: „Die Inverkehrbringung solcher Sets als Behandlungseinheit nach Artikel 22 MDR ist nur möglich, sofern die darin befindlichen Produkte in ihrem jeweiligen bestimmungsgemäßen Gebrauch auch für die Verwendung durch Laien vorgesehen sind.“
-> Muss die Verwendung durch Laien explizit in der Zweckbestimmung bzw. dem erweiterten bestimmungsgemäßen Gebrauch vorgesehen sein? Zum Beispiel, wenn der Hersteller eines Alkoholtupfers in seiner Zweckbestimmung schreibt „Verwendung als antiseptisches Hautreinigungsmittel vor Injektionen und Blutentnahmen“ und keine weitere User Gruppe angibt, dann kann doch angenommen werden, dass der Hersteller ALLE Menschen einbezieht, womit die Gruppe der Laien auch enthalten sind. Ansonsten müsste der Hersteller den Verkauf des Alkoholtupfer an Laien explizit ausschließen, damit Apotheken etc. diese nicht an die Gruppe herausgeben. Ich habe zum Beispiel des öfteren Medizinprodukte gesehen, die die User Gruppe spezifieren („Layperson“ oder „Healthcare professional“), aber auch viele, die es nicht machen.
2. Sie schreiben:
„War eine Verwendung der Bestandteile des Sets durch Laien nicht vorgesehen, muss das Probenahme-Set als CE-markiertes Produkt zur Eigenanwendung in Verkehr gebracht werden. Somit würde das Set gemäß IVDR, Anhang VIII, Regel 4 mindestens in Klasse C fallen. […] Erfüllen alle Bestandteile des Sets die Anforderungen an eine Eigenanwendung durch Laien, kann das Set dennoch nur als Behandlungseinheit nach MDR Artikel 22 in Verkehr gebracht werden, wenn der Laie nach der Probenahme keine Tätigkeiten durchführen muss, die die Schwelle zum Selbsttest überschreiten.“
Grunsätzlich müsste man bei DTC ebenfalls zwischen MDR und IVDR Sets unterscheiden.
-> Ein „Produkt zur Eigenanwendung“ (IVD) ist laut dem Borderline (Kap. 2.1, Vers, 1.22, 2019) nur anwendbar, wenn das Produkt „verwendet“ wird (IVDR, Art.2 (5)) und ein „Self-Testing“ als Verwendung einschließt. Das „testing“ beinhaltet hier ein Resultat oder zumindestens etwas, dass der Patient vor dem Versand der Probe unmittelbar mit der Probe tun muss, um den Begriff „used“ zu erfüllen. Das bedeutet, ein IVD Probengefäß (z.B Röhrchen), welches einfach nur der Sammlung der Probe dient, ist niemals ein „Produkt zur Eigenanwendung“ und eine DTC-Test, der nur die Zweckbestimmung hat, Probenmaterial zusammeln und zum Labor zu schicken, wird demnach auch nie ein „Produkt zur Eigenanwendung“ darstellen, außer der Kunde erhält unmittelbar mit dem Set ein Ergebnis oder er muss eine Art Präanalyse durchführen. Diese Abweichung ergibt sich aus dem deutschen Wort „Eigenanwendung“ und dem englischen „Self-testing“. Das Borderline Dokument beschreibt dieses Problem sehr gut. Sie haben das selber auch geschrieben: “ […] der Laie nach der Probenahme keine Tätigkeiten durchführen muss, die die Schwelle zum Selbsttest überschreiten.“
a) Demzufolge müsste ein DTC-Test, der ausschließlich dem Zweck der Probenentnahme dient und nicht nach MDR Art. 22 einklassifiziert werden kann (z.B. expliziter Auschluss von Laien in der Anwendung in mind. einer Komponente), eher gemäß IVDR, Anhang VIII Klasse B (Regel 6) eingeordnet werden.
b) Für Behandlungseinheiten nach MDR Art. 22 ist es sogar noch komplizierter, denn die MDR spezifiert den Bergriff „Eigenanwendung“ gar nicht. Hier gibt es nur den „Anwender“. Zum Beispiel könnte ein DTC-Test, der nur aus MDR-Komponenten besteht, wobei ein Art. 22 MDR nicht zulässig ist (Anwendung durch Laien explizit ausgeschlossen), ebenfalls nicht als „Produkt zur Eigenanwendung“ gemäß IVDR klasifiziert werden. Zum Beispiel kenne ich Abstrichtupfer, die ohne extra Probenentnahemröhrchen vertrieben werden. Diese Tupfer werden durch die Hersteller unter der MDR und nicht IVDR klassifiziert, obwohl die getrocknete Probe auf dem Tupfer verbleibt.
-> Damit wären auch DTC-Test als Behandlungseinheiten oder eigenständiges Medizinprodukte nach MDR ohne ein explizites IVD Probenröhrchen möglich.
Sehr geehrter Herr Salis,
ich danke Ihnen sehr für Ihre präzise Analyse.
Beginnen wir mit 2.
Zu 2. a):
Sie haben recht in Ihrer Argumentation. Das Borderline Manual sagt „Thus where a specimen receptacle is simply used by the patient to contain a specimen it remains a specimen receptacle“. Somit muss ein Probengefäß bei einfacher Probenahme durch den Laien, ohne weitere nachfolgende Tätigkeiten (außer dem Versand), kein Produkt zur Eigenanwendung sein.
Damit kann ein DTC-Test Probenahme-Set theoretisch auch als Produkt der Klasse B in Verkehr gebracht werden. Ich habe den Artikel entsprechend angepasst.
Bleibt noch für die Zukunft offen, wie das aktuelle Borderline Manual zukünftig solch eine Situation interpretiert.
Zu 2. b):
Ein DTC-Test Probenahme-Set, das nur aus MDR-Komponenten besteht, kann es nicht geben. Denn wie in IVDR, Artikel 2 2. steht ist ein Probenbehältnis ein IVD. Wenn der von Ihnen beschriebene Abstrichtupfer zur Probenaufnahme und -Transport genutzt wird, ist er ein Probenbehältnis nach IVDR, Artikel 2 3. und muss entsprechende Anforderungen erfüllen. Ist das nicht der Fall ist das Produkt regulatorisch nicht konform und illegal in Verkehr gebracht.
Zu 1.:
Korrekt, bei Medizinprodukten nach MDR ist nicht immer sofort ersichtlich, ob das Produkt für die Anwendung durch Laien bestimmt ist. Auch sind die zusätzlichen Anforderung der MDR an solche Produkte weitaus geringer im Vergleich zu den zusätzlichen Anforderungen der IVDR an IVD zur Eigenanwendung gegenüber ihrem Pendant für den professionellen Gebrauch. In der MDR sind diese Anforderungen insbesondere in Anhang I 22. zu finden und resultieren u. a. in besonderen Aufwänden bei der Gebrauchstauglichkeit. Ist es aus der Gebrauchsanweisung des Medizinproduktes nicht ersichtlich, ob es für die Anwendung durch Laien bestimmt ist und die Anforderungen aus Anhang I 22. erfüllt sind, sollten man dies beim Hersteller erfragen.
Sehr geehrter Herr Hafen,
zu 1) Das ist stimmt, sofern der Hersteller diese Informationen aus der technischen Dokumentation bereitstellt.
zu 2a) Auf das neue Borderline Dokument bin ich auch gespannt.
zu 2b) Generall würde ich Ihnen zustimmen und ich sehe die Einstufung auch ähnlich. Aber für die invasiven Abstrichtupfer OHNE zusätzliches Probennahmeröhrchen würde ich Ihnen leider widersprechen. Nach dem MEDDEV „GUIDELINES ON MEDICAL DEVICES – IVD Medical Device Borderline and Classification issues“ (2.14/1 rev.2, 01/2012) wird unter Punkt „1.3.2. Products used to obtain specimens“ folgendes geschrieben:
„b) With intended direct contact with the human body
Invasive sampling devices or those which are directly applied to the human body for the purpose of obtaining a specimen within the meaning of Directive 93/42/EEC shall not be considered to be accessories to in vitro diagnostic medical devices (Art 1.2.c) (e.g. needles, lancets, lancing devices, mouthtubes, swabs, urine collection bags for babies). These products are regarded as being devices within the scope of Directive 93/42/EEC.“
Unter Kapitel “ 1.3.1. Specimen receptacles“ wird nochmal deutlich der Aspekt von invasiven Medizinprodukten hervorgehoben: „Specimen receptacles with invasive body contact should be handled in the same way as described in chapter 1.3.2.b.“
Viele Hersteller für Swabs (ohne weiteres IVD Röhrchen) klassifizieren die Produkte unter Annex VIII, Rule 5 (INVASIVE DEVICES – All invasive devices with respect to body orifices, other than surgically invasive devices).
Problem: Das Borderline bezieht sich leider noch auf die MDD/IVDD und nicht MDR/IVDR. Aber da es (noch) kein aktualisiertes Borderline Dokument für die MDR/IVDR gibt, spiegelt das alte Borderline der MEDDEV ebenfalls den Stand der Technik , da die Definition sich zwischen der MDR/IVDR und MDD/IVDR sich nicht grundlegend geändert haben.
Würden Sie das auch so sehen? Vielen Dank für die Auskunft.
Lieber Herr Salis,
vielen Dank für Ihr Nachhaken.
In MEDDEV 2.14/1 1.3.2. b) geht es um Produkte mit dem Zweck „obtaining a specimen“. Es geht also rein um die Gewinnung der Probe, nicht um die Aufbewahrung und Lagerung, wie bei einem Probenbehältnis.
Eine Ausnahme bilden in der Tat invasive Produkte, wie z. B. auch Swabs zur Gewinnung von Nasensekret. Auf diese trifft der von Ihnen zitierte Absatz aus MEDDEV 2.14/1 1.3.1. klar zu. Dennoch komme ich hier in einem Probenahme-Set nicht ohne ein weiteres Probenbehältnis wie ein Probenröhrchen aus, in das ich das Swap oder den invasiven Abstrichtupfer gebe, schon alleine um die Integrität der Probe bei der Lagerung und dem Transport sicherzustellen. Dies sollte spätestens als Maßnahme aus dem Risikomanagement umgesetzt werden.
Es gibt bereits ein auf IVDR und MDR bezogenes Borderline Manual, doch leider aktuell noch sehr lückenhaft befüllt, sowie ein paar weitere hilfreiche aktuelle Dokumente (siehe hier). Dennoch können/müssen das „alte“ Borderline Manual in teilen sowie manche MEDDEV Dokumente noch als Stand der Technik herangezogen werden.
Gibt es weiteren Klärungsbedarf, melden Sie sich doch gerne direkt bei mir.