
Eine Person Responsible for Regulatory Compliance (PRRC) fordern sowohl die MDR als auch die IVDR.
Manche sprechen auch von einer Artikel-15-Person (nach den entsprechenden Artikeln in den beiden EU-Verordnungen). Im Deutschen nutzt man die Formulierungen der für die Einhaltung der regulatorischen Vorschriften verantwortlichen Person (kurz: verantwortliche Person) bzw. der qualifizierten bzw. der sachkundigen Person.
Die Person Responsible for Regulatory Compliance hat Ähnlichkeiten mit den Sicherheitsbeauftragten, ist aber damit nicht deckungsgleich.
Erfahren Sie, welche Aufgaben die PRRC ausführen, welche Verantwortungen sie übernehmen muss und welche Kompetenzen zwingend sind. So können Sie Ordnungsstrafen von bis zu 30.000 Euro vermeiden.

Mit dem Laden des Videos akzeptieren Sie die Datenschutzerklärung von YouTube.
Mehr erfahren
1. Regulatorische Anforderungen an die PRRC
a) MDR und IVDR
Motivation
Den Autoren der MDR (Medical Device Regulation) war wichtig, dass es eine Person Responsible for Regulatory Compliance gibt, um u. a. die Herstellung und das Marktüberwachungs- und Meldesystem zu verbessern. Sie schreiben in der Präambel:
It should be ensured that supervision and control of the manufacture of and the post-market surveillance and vigilance activities of medical devices are carried out within the manufacturer’s organisation by a person responsible for regulatory compliance who fulfils minimum conditions of qualification.
„Verantwortliche Person“ ist verpflichtend
Folgerichtig fordert die MDR in Artikel 15 solch eine „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“. Die nahezu wortgleiche Forderung stellt die IVDR (ebenfalls in Artikel 15). Beide Artikel legen fest:
- Aufgaben der verantwortlichen Person (Absatz 3)
- Kompetenzen der verantwortlichen Person (Absatz 1) sowie die Anforderungen an deren Nachweis (Absatz 6)
- Ausnahmen, unter denen die verantwortliche Person nicht Teil der Organisation sein muss (Absatz 2)
Die EU hat einen Leitfaden Guidance on Article 15 MDR-IVDR Person responsible for Regulatory Compliance veröffentlicht. Für 2021 ist dessen Überarbeitung geplant. Sie finden hier die Roadmap.
Registrierung ist verpflichtend
Der Artikel 31 in Verbindung mit dem Anhang VI, Teil 1, Absatz 1. bzw. 1.4 besagt, dass „die Hersteller oder gegebenenfalls ihre Bevollmächtigten und, sofern zutreffend, die Importeure „Name, Anschrift und Kontaktdaten der für die Einhaltung der Regulierungsvorschriften zuständigen Person(en) gemäß Artikel 15“ in der EUDAMED registrieren müssen.
Auch das BfArM weist darauf hin, dass die PRRC im Rahmen der Registrierung der Wirtschaftsakteure im Actor Registration Module angegeben werden muss.
Beachten Sie: Die Forderung nach einer „Artikel-15-Person“ betrifft nicht nur die Hersteller, sondern auch die Bevollmächtigten!
Im oben genannten Guidance Dokument betont die EU, dass die für die Einhaltung der Regulierungsvorschriften verantwortliche Person NICHT gleichzeitig verantwortliche Person beim Hersteller und beim EU-Beauftragten (Authorized Representative) sein kann.
Die verantwortlichen Personen muss eine Nähe zum Hersteller haben, d. h. bei einem EU-Hersteller innerhalb der EU ansässig sein und bei einem Nicht-EU-Hersteller außerhalb der EU.
b) Nationales Recht und Strafen
In Deutschland gilt seit dem 25.05.2021 das Medizinproduktedurchführungsgesetz MPDG. Dieses stellt keine zusätzlichen, über die MDR hinausgehenden Anforderungen an die „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“.
Das MPDG bestimmt allerdings in § 94 Absatz 3 die Höhe des Bußgeldes (bis zu „dreißigtausend“ (sic!) Euro. Dieses kann fällig werden, falls:
„a) entgegen Artikel 15 Absatz 1 in seiner Organisation nicht über eine oder nicht über eine danach qualifizierte Person, die für die Einhaltung der Regulierungsvorschriften verantwortlich ist, verfügt,
MPDG § 94
b) entgegen Artikel 15 Absatz 2 nicht auf eine qualifizierte Person, die für die Einhaltung der Regulierungsvorschriften verantwortlich ist, ständig und dauerhaft zurückgreifen kann,
c) entgegen Artikel 15 Absatz 6 nicht auf eine danach qualifizierte Person, die für die Einhaltung der Regulierungsvorschriften verantwortlich ist, ständig und dauerhaft zurückgreifen kann“
Die Rolle des Sicherheitsbeauftragten hat das MPDG abgeschafft, die des Medizinprodukteberaters beibehalten.
2. Aufgaben und Verantwortlichkeiten der veranwortlichen Person
a) Verantwortlichkeiten
Laut MDR und IVDR sind die responsible persons dafür verantwortlich(!), dass Folgendes sichergestellt wird:
- Konformität der Medizinprodukte wird in Übereinstimmung mit dem QM-System (vor deren Auslieferung) geprüft (Artikel 10(9)).
- Technische Dokumentation wird aktuell gehalten [Artikel 10(4) und (6)].
- Die Pflichten zur Überwachung nach dem Inverkehrbringen erfüllt werden gemäß [Artikel 10(10)].
- Meldepflichten werden gemäß den EU-Richtlinien erfüllt [Artikel 10(13)].
- Bei Investigational Devices wird die Erklärung gemäß Anhang XV, Kapitel 2 ausgestellt.
Diese Liste listet auch die Referenzen zu Artikel 10, die die EU im Guidance on Article 15 MDR-IVDR Person responsible for Regulatory Compliance nennt.
Weder die MDR noch die IVDR fordern, dass die „persons responsible for regulatory compliance“ die Aufgaben selbst durchführen müssen. Sie sind „lediglich“ für deren Erledigung verantwortlich. Die damit einhergehende Verantwortlichkeit muss aber auch zu einem disziplinarischen Einfluss führen.
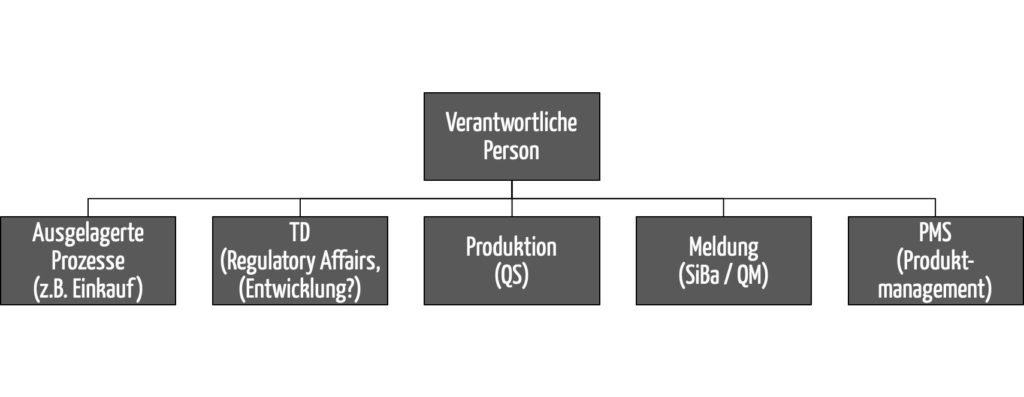
b) Abgrenzungen
Abgrenzung zum QM-Beauftragen
Die Aufgaben der verantwortlichen Person und des QM-Beauftragten (QMB) überlappen sich. Eine Gegenüberstellung findet sich im Artikel zum QM-Beauftragten.
Abgrenzung zum Sicherheitsbeauftragten
Mit der MDR und dem MPDG sind das MPG und dessen Anforderungen an Sicherheitsbeauftragte weggefallen. Daher ist dieses Teilkapitel obsolet.
30 MPG Absatz 4 fordert von den Sicherheitsbeauftragten:
„Der Sicherheitsbeauftragte für Medizinprodukte hat bekannt gewordene Meldungen über Risiken bei Medizinprodukten zu sammeln, zu bewerten und die notwendigen Maßnahmen zu koordinieren. Er ist für die Erfüllung von Anzeigepflichten verantwortlich, soweit sie Medizinprodukterisiken betreffen.“
Die Aufgaben und Verantwortlichkeiten eines Sicherheitsbeauftragten und einer verantwortlichen Person (PRRC) sind somit nicht identisch:
| Aufgabe/Verantwortlichkeit | MDR/IVDR | MPG |
|---|---|---|
| Prüfung der Konformität der Medizinprodukte | X | — |
| Technische Dokumentation (auch deren Aktualität) | X | — |
| Marktüberwachung | X | X |
| Meldewesen | X | X |
c) Haftung der verantwortlichen Person (PRRC)
Die verantwortliche Person haftet in Fällen einfacher Fahrlässigkeit in der Regel nicht persönlich. Bei grober Fahrlässigkeit kann der Hersteller seine:n Mitarbeiter:in in Regress nehmen.
Eine Haftungsbeschränkung oder eine Freistellungsabrede kann in den Arbeitsvertrag mit der verantwortlichen Person aufgenommen werden.
Bei grober Fahrlässigkeit oder Vorsatz ist ein solcher Ausschluss nicht sinnvoll und möglich. Zudem drohen strafrechtliche Konsequenzen.
Die verantwortliche Person darf aufgrund ihrer Pflichten nicht benachteiligt werden.
Lesen Sie hier unseren ausführlichen Beitrag zur Arbeitnehmerhaftung.
Der englische Begriff responsible person lässt sich ins Deutsche auch mit zuständige Person übersetzen. Das klingt schon etwas freundlicher. Schließlich geht es nicht darum, einen Schuldigen zu finden.
Danke an Dr. T. Castner für diesen Gedanken.
3. Kompetenzen
a) Anforderungen an die Kompetenzen
Die Richtlinien fordern, dass die Kompetenz der verantwortlichen Person nachgewiesen wird. Für diesen Nachweis sehen sie zwei Varianten vor:
- Universitätsabschluss oder anerkannter Studienabschluss in Jura, Medizin, Pharmazie, Engineering oder einer anderen relevanten wissenschaftlichen Disziplin UND mindestens ein Jahr Berufserfahrung in den Bereichen Qualitätsmanagement oder Regulatory Affairs. Diese Berufserfahrung muss sich auf Medizinprodukte beziehen.
- Vier Jahre Berufserfahrung in den Bereichen Qualitätsmanagement oder Regulatory Affairs. Diese Berufserfahrung muss sich auf Medizinprodukte beziehen.
Bei Herstellern von Sonderanfertigungen erlauben die EU-Verordnungen, den Nachweis auf nur zwei Jahren Berufserfahrung zu verringern.
b) Anforderungen an den Nachweis
Die Studienabschlüsse sind eindeutig zu belegen. Bei der Berufserfahrung sollten die Hersteller einen oder mehrere der folgenden Nachweise erbringen können:
- Arbeitsvertrag, aus dem hervorgeht, dass und seit wann die Person im Bereich Regulatory Affairs oder Qualitätsmanagement arbeitet
- Organigramm, die diese Rolle erkennen lässt
- Schulungsnachweise zu den Themengebieten Qualitätsmanagement, Medizinprodukterecht, Marktüberwachung, Risikomanagement, Meldewesen
- Aufzeichnungen wie Auditberichte und Dokumentenfreigaben
- Meldung der Person beim DIMDI als Sicherheitsbeauftragter
Beachten Sie, dass neben den Anforderungen der MDR auch die der ISO 13485 zu erfüllen sind, die seit der Version 2016 die Kompetenz noch stärker in den Fokus rückt.
c) Vergleich mit den Kompetenzen von Sicherheitsbeauftragten
Die von den Sicherheitsbeauftragten geforderten Nachweise unterscheiden sich in den folgenden Punkten von denen der verantwortlichen Person:
- Jura zählt nicht zu den anerkannten Hochschulabschlüssen.
- Unabhängig von einem Studium sind immer mindestens zwei Jahre Berufserfahrung gefordert.
- Die Berufserfahrung bezieht sich auf den etwas enger definierten Aufgabenbereich des Medizinproduktegesetzes. Bei den EU-Richtlinien zählt auch Berufserfahrung im Qualitätsmanagement.
4. PRRC als externe Kompetenz?
Hersteller müssen eine PRRC innerhalb ihrer Organisation ansiedeln. Die MDR erlaubt Ausnahmen für Kleinst- und Kleinunternehmen. Aber auch in diesen Fällen müssen sie „dauerhaft und ständig auf eine solche Person zurückgreifen können.“ Die EU macht in ihrem Leitfaden Guidance on Article 15 MDR-IVDR Person responsible for Regulatory Compliance klar, dass dieser Zugriff vertraglich geregelt sein muss.
Kleinste und kleine Organisationen dürfen sich externer Personen bedienen. Sie sind von der Pflicht befreit, selbst eine PRRC anzustellen.
Was eine kleine bzw. kleinste Organisation ist, definiert die EU in der Richtlinie 2003/361/EC: Demnach zählen Firmen mit weniger als 50 Mitarbeitenden und einer Jahresbilanz von maximal 10 Mio. Euro dazu.
Insbesondere für Kleinst- und kleine Unternehmen übernehmen wir gerne die Rolle der verantwortlichen Person gemäß MDR und IVDR und sichern Ihre regulatorische Compliance.
5. Übergangsfristen
Seit Gültigkeitsbeginn der MDR gibt es Uneinigkeit darüber, ob auch für „Legacy-Produkte“ eine „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“ gemäß MDR Artikel 15 benannt werden muss.
Am 19. Oktober wurde die MDCG Guidance 2021-25 veröffentlicht. Dieses Guidance Dokument interpretiert und konkretisiert Anforderungen an QM-Systeme von Herstellern, die nur „Legacy Devices“ in Verkehr bringen.
Legacy devices should be understood as devices, which, in accordance with Article 120(3) of the MDR, are placed on the market after the MDR’s date of application (DoA) and until 26 May 2024 if certain conditions are fulfilled. Those devices can be:
- devices which are class I devices under Directive 93/42/EEC (MDD), for which an EC declaration of conformity was drawn up prior to 26 May 2021 and for which the conformity assessment procedure under the MDR requires the involvement of a notified body;
- devices covered by a valid EC certificate issued in accordance with Directive90/385/EEC (AIMDD) or the MDD prior to 26 May 2021
Quelle: MDCG 2021-25
Inhalt des Dokuments
Solange ein Hersteller nur Produkte nach der Übergangsbestimmung Art. 120 (3) in Verkehr bringt, also noch keine Produkte nach der MDR CE-kennzeichnet, wird noch keine verantwortliche Person nach Art. 15 benötigt.
Bisher hatten wir und auch viele andere dies anders interpretiert. Der Grund dafür war, dass die Pflichten für die Post-Market Surveillance (PMS) und die Vigilanz ab dem 26.05.2021 von allen Herstellern erfüllt sein müssen. Die verantwortliche Person ist für einen wirksamen PMS- und Vigilanzprozess verantwortlich. Die Guidance MDCG 2021-25 betrachtet diesen Aspekt auch, kommt aber zu dem Schluss, dass Artikel 15 MDR noch nicht auf Hersteller von Legacy-Produkten anzuwenden ist.
Eine ähnliche Aussage trifft MDCG 2022-8 für die IVDR. Im Anhang stellt dieses Dokument ebenfalls fest, dass Artikel 15 IVDR (Person Responsible for Regulatory Compliance) für Legacy-Produkte noch nicht gilt.
Das wiederum unterstützt die Interpretation, dass die verantwortliche Person für „supervision and control“ zuständig ist und eben nicht alles selbst machen muss (denn sonst könnte man kaum die Meinung vertreten, dass Hersteller von Legacy-Produkten keine verantwortliche Person benötigen.)
Bitte beachten Sie: Alle Hersteller müssen in jedem Fall wirksame Prozesse für Post-Market Surveillance und Vigilanz implementieren. Nur die Überwachung durch die verantwortliche Person ist noch nicht zwingend für Hersteller von Legacy-Produkten vorgeschrieben.
6. Zusammenfassung
Die Anforderungen an die verantwortliche Person (PRRC) unterscheiden sich nach Art und Größe des Wirtschaftsakteurs bzw. der Rolle.
| Großunternehmen | Kleinunternehmen | Kleinstunternehmen | Sonderanfertiger | Bevollmächtigter | |
| Mitarbeiter | ≥ 50 | < 50 | < 10 | – | – |
| Jahresumsatz | > 10 Mio. € | ≤ 10 Mio. € | ≤ 2 Mio. € | – | – |
| Intern/ extern | Interne VP |
Zugriff auf eine VP |
Zugriff auf eine VP | Zugriff auf eine VP |
Zugriff auf eine VP |
| Qualifikation | Hochschulzeugnis + 1 Jahr Berufserfahrung in QM/RA | Hochschulzeugnis + 1 Jahr Berufserfahrung in QM/RA | Hochschulzeugnis + 1 Jahr Berufserfahrung in QM/RA | – | Hochschulzeugnis + 1 Jahr Berufserfahrung in QM/RA |
| oder | 4 Jahre Berufserfahrung in QM/RA | 4 Jahre Berufserfahrung in QM/RA | 4 Jahre Berufserfahrung in QM/RA | 2 Jahre Berufserfahrung in der Fertigung | 4 Jahre Berufserfahrung in QM/RA |
| Benachteiligung | Keine | Keine | Keine | Keine | – |
MDR und IVDR haben mit den „für die Einhaltung der regulatorischen Anforderungen verantwortlichen Personen“ eine Rolle eingeführt, deren Verantwortung und Bedeutung weit über die des Sicherheitsbeauftragten hinausgeht.
Hersteller sind gut beraten, diese Rollen schnellstens qualifiziert zu besetzen, um
- die regulatorischen Anforderungen zu erfüllen,
- die Sicherheit und Konformität ihrer Produkte zu gewährleisten und
- schmerzhafte Strafen zu vermeiden.
Änderungshistorie
- 2023-08-11: Abgrenzung zum QMB eingefügt. Hinweis ergänzt, dass MPG nicht mehr gültig ist.
- 2022-08-24: Link zu MDCG 2022-8 (IVDR Legacy Devices) ergänzt
- 2021-06-14: Artikel redaktionell überarbeitet und um Kapitel mit Übergangsfristen ergänzt
- 2021-11-19: Abschnitt 5 in Bezug auf MDCG Guidance 2021-25 (Autor Alexander Thern) überarbeitet



Die MDR fordert die „Person responsible for regulatory compliance“ im Artikel 15, nicht 13.
Sie haben absolut Recht! Wird sofort geändert.
Worauf basiert Ihre Aussage zur Haftung der Person responsible for Regulatory Compliance?
Sehr geehrte Frau S.,
die Aussage zur Haftung folgt aus einer Analogiebildung mit der Haftung von Sicherheitsbeauftragten. Diese findet sich allerdings nur teilweise im Gesetz (z.B. BGB). Meist ist es „gesprochenes Recht“.
Wir werden noch einen umfassenden Beitrag zur Arbeitnehmerhaftung publizieren. Danke für Ihre Anregung dazu!
Beste Grüße, Christian Johner
Hallo!
Es ist meinerseits nicht klar definiert, ob der Responsible f. R. C im legal Hersteller sitzen muss, oder auch im EC-REP vertreten sein kann.
Wie sehen Sie das?
Ansonsten gute Zusammenfassung. Danke dafür!
LG
DS
Sehr geehrter Herr S.,
danke für die wichtige Frage: Die „Artikel-15-Person“ kann bei kleinen Firmen (SMEs) ausgelagert werden. Das kann auch an der EC-REP sein.
Artikel 11 (zum EC-REP) sagt: „Der Bevollmächtigte führt die Aufgaben aus, die in dem zwischen ihm und dem Hersteller vereinbarten Mandat festgelegt sind.“. Die Delegation der Aufgabe gemäß Artikel 15 ist nicht ausgeschlossen: „(4) Das in Absatz 3 des vorliegenden Artikels genannte Mandat kann nicht die Pflichten des Herstellers gemäß Artikel 10 Absätze 1, 2, 3, 4, 6, 7, 9, 10, 11 und 12 delegieren.“
Damit ist die Antwort, dass die Delegation an den EC-REP möglich ist.
Viele Grüße, Christian Johner
Eine kurze Frage zum Thema qualified Person:
Wie verhält es sich mit der kündbarkeit einer qualified Person ?
Mit freundlichen Grüßen
Jack
Das Kündigungsrecht können nur die Nationalstaaten regeln. Die MDR sagt aber, dass die Person keine Nachteile erleiden darf. Daher würde ich von einer etwas höheren Hürde ausgehen.
Hallo Herr Johner,
Bzgl. ihrer Antwort an Herrn S hinsichtlich der Frage ob die „Person Responsible for Regulatory Compliance“ (PRRC) an den EC-Rep ausgelagert werden kann, würde ich gerne einen ergänzenden Beitrag hinzufügen.
Grundsätzlich hat der EC-Rep eine eigene PRRC zu benennen. Diese PRRC EC-Rep ist verantwortlich dafür, dass der EC-Rep seine Verantwortlichkeiten wie in Artikel 11 beschrieben erfüllt.
Lagert der Hersteller die PRRC Mfr. aus, sind die spezifischen Herstellerverantwortlichkeiten zu berücksichtigen.
Sollte also der EC-Rep die PRRC Mfc. stellen, ist dies eine zusätzliche Dienstleistung, losgelöst von der Funktion EC-Rep und es sind die unterschiedlichen Verantwortlichkeiten der PPRC EC-Rep und der PRRC Mfr. getrennt festzulegen. Insbesondere der Hersteller hat dies in seinem QM-System zu dokumentieren.
Die PRRC Mfr ist nie identisch mit der PRRC EC-Rep, da die PRRC EC-Rep. nie Aufgaben übernehmen kann, die über die Verantwortlickeiten des EC-Rep hinausgehen. So beschreibt Artikel 15(3) Aufgaben der PRRC, die nicht an den EC-Rep delegiert werden können (z.B. Überprüfung der Konformität gemäß QM im Rahmen der Produktion)
Der Hersteller kann also nicht einfach (in seinem QM-System) beschreiben, dass die Funktion der PRRC an den EC-Rep delegiert ist. Es sollten auch zwei Verträge (Mandate) abgeschlossen werden. Zum einen mit dem EC-Rep und zum anderen mit der Person, die die Funktion PRRC Mfr übernimmt.
Ich hoffe ich konnte meinen Punkt klar machen. Da es hierzu noch keine einheitliche Aussage gibt, ist natürlich abzuwarten, was von den Benannten Stellen und den Zuständigen Behörden akzeptiert wird.
Mfg
Clemens Mohr
Unter den Verantwortlichkeiten, Punkt 5, hat sich ein kleiner Fehler eingeschlichen. Es sollte heißen: Bei „Investigational Devices“ wird die Erklärung gemäß Anhang XV, Kapitel 2 ausgestellt.
Viele Grüße
MK
Sie haben Recht, liebe Frau Kotlyarov!
Danke für Ihren Hinweis, dank dessen ich die Nummer sofort korrigieren werde. Da habe ich ein „X“ mit einen „I“ verwechselt.
Nochmals besten Dank!
Viele Grüße, Christian Johner
Hallo Herr Johner,
wenn ich den Artikel richtig verstanden habe, dann werden die beiden Begriffe „Für die Einhaltung der Regulierungsvorschriften verantwortliche Person“ und die „Qualified Person“ synonym verwendet?!
Ist es korrekt, dass der QMB eines Unternehmens nicht zeitgleich auch als Qualified Person auftreten darf?
Beste Grüße
MS
Welcher Begriff sich für die „Artikel-15-Person“ durchsetzen wird, ist noch unklar. Wir halten „Qualified Person“ für eine gute Option.
Es gibt keine Vorschrift, die es unterbindet, dass der QMB auch die Qualified Person ist.
Sehr geehrter Herr Prof. Johner,
ich habe folgende Frage gleich zum ersten Satz des Artikel 15 (1) MDR.
Er fordert explizit nur die Einsetzung eines PRRC für „Hersteller“.
Sind Importeure und Händler somit ganz von diesem Artikel ausgenommen und müssen somit kein PRRC bestellen?
Ich nehme an, dass bei den in Art. 15 (2) genannt Kleinst- und Kleinunternehmen auch „Hersteller“ gemeint sind, obgleich nicht direkt erwähnt?
Grund meiner Frage sind folgende Ähnlichkeiten im Verantwortungsbereich des PRRC und des Importeurs:
1- der Importeur muss nach Art. 13 MDR ja auch an die Behörde melden (auch Rückruf), wenn eine schwerwiegende Gefahr von einem Produkt ausgeht. (Art. 13 (2) ähnelt also den Berichtspflichten wie in Art. 15 (3d))
2-der Importeur muss im Rahmen seiner Möglichkeiten die Konformität der Produkte sicherstellen (Art. 13, dort im wesentlichen: (2a), (2c), (7) ähnelt somit Art. 15 (3a) und (3b))
Mit freundlichen Grüßen
Uwe Boetcher
Sehr geehrter Herr Boetcher,
die Pflichten richten sich in der Tat an die Hersteller bzw. an die EU-Bevollmächtigten. Das macht auch Sinn, weil die Händler und Importeure oft nicht über dieses Fachwissen verfügen.
Wie gesagt, bei einem Import muss der EU-Bevollmächtigte die Anforderungen des Artikels 15 (Absatz 6) erfüllen.
Falls die Händler und Importeure jedoch die in Artikel 16 genannten Tätigkeiten zu übernehmen, gelten für sie die Herstellerpflichten — auch die des Artikels 15.
Ihre Beobachtungen bezüglich der Ähnlichkeiten sind zutreffend. Eine Teilmenge der Verantwortlichkeiten einer PRRC übernehmen sie. Der Artikel 15 wendet sich dennoch nur an Hersteller und Bevollmächtigte.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
im Artikel hieß es, dass die Qualified Person nicht mit dem Sicherheitsbeauftragten gleich zu setzen ist. Wie ist das in personeller Hinsicht? Braucht es somit in einem Betrieb jeweils eine Person für jeh eine der genannten Position, oder wie soll das gehandhabt werden? Mir ist bewusst, dass die Tätigkeiten der QP auf mehrere Personen aufgeteilt werden können, braucht es dennoch eine Benennung eines Sicherheitsbeauftragten?
In Ihrem Artikel zu den Aufgaben des Sicherheitsbeauftragten wird davon gesprochen, dass die Aufgaben mit der MDR erweitert werden sollen und das sich auch die Bezeichnung noch ändern wird. Kann das so verstanden werden, dass der Sicherheitsbeauftragte durch mehr Aufgaben zur QP werden soll? Habe ich hier vielleicht etwas falsch aufgefasst?
Mit freundlichen Grüßen,
Daniel Planteu
Sehr geehrter Herr Planteu,
das Medizinproduktedurchführungsgesetzt MDG, der Nachfolger des MPG, sieht keinen Sicherheitsbeauftragten mehr vor. Damit erübrigt sich die Frage, ob das die gleiche Frage sein soll.
Allerdings liegt das MDG erst im Referentenentwurf vor.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
wenn die Aufgaben der QP auf mehrere Personen aufgeteilt werden, dies klar beschrieben wird und im Organigramm dargestellt ist, wie ist dann die Hinterlegung der Daten bei Behörden etc.? Als aktuelles Beispiel, welcher QP wird in der EUDAMED angegeben? Wer ist der Ansprechpartner für Behörden? Müssen alle QPs angegeben werden oder gibt es einen „übergeordneten“ QP?
Mit freundlichen Grüßen
G. Graubner
Sehr geehrte Frau Graubner,
laut 1.4 in Anhang VI müssen alle Personen, die für die Einhaltung verantwortlich sind, eingetragen werden. Daher spricht die MDR von „Person(en)“. Das Konzept einer „übergeordneten QP“ ist mir nicht bekannt. Ob die EUDAMED eine Identifizierung eines primären Ansprechpartners ermöglichen wird, werden wir sehen. In der derzeitigen Testphase ist das noch nicht erkennbar.
Beste Grüße, Christian Johner
Hallo Herr Johner
Es ist zu befürchten, dass die Schweiz in Kürze zum Drittstaat wird.
Wäre dann folgendes möglich:
Ein MP-Hersteller befindet sich in der Schweiz (z.B. in Kreuzlingen).
Die Hersteller-PRRC ist Mitarbeiter des Herstellers.
Der EC REP (gleichzeitig IMP) befindet sich in Deutschland (z.B. in Konstanz).
Kann die Hersteller-PRRC gleichzeitig die PRRC für den EC REP sein?
Die geforderte Nähe zum EC REP wäre vorhanden.
Ich finde in der MDR und auch im MDCG 2019-7 nichts, das dagegen spricht.
Wie sehen Sie das?
MfG
JS
Sehr geehrter Herr Schneider,
es ist nicht nur zu befürchten, sondern sogar relativ wahrscheinlich, dass die Schweiz in Kürze ein Drittstaat wird, was natürlich das Herz bluten lässt.
Ich sehe auch nichts, was gegen Ihren Vorschlag spricht.
Mit herzlichen Grüßen an die heimatliche Grenze, Christian Johner
Sehr geehrter Herr Johner,
ich bin beim Lesen des (hervorragenden) Artikels über den Teil gestolpert, dass eine Qualified Person auch für Importeure benötigt wird.
Ich habe mir die drei Stellen in der MDR angesehen und interpretiere hier ein wenig anders.
In Artikel 15 wird die Qualified Person für den Hersteller gefordert. Auch die in Artikel 15 beschriebenen Aufgaben passen zu denen eines Herstellers und nicht zu denen eines Importeurs. Ich interpretiere den Anhang, auch in Anbetracht der Worte „sofern zutreffend“ vor den Importeuren, dass die Forderung hier nicht für die Importeure, sondern nur für die Hersteller und die Bevollmächtigten gilt.
Haben Sie hierzu noch weitere Informationen oder Interpretationen? Gerne lasse ich mich von Ihnen überzeugen, auch wenn ich diese Forderung deer MDR für nicht sinnvoll halte.
Mit besten Grüßen,
Robert Moldenhauer
Sehr geehrter Herr Modlenhauer,
Sie haben Recht, das ist nicht ganz präzise. Das sofern zutreffend bezieht sich (wahrscheinlich) auf Artikel 16, in denen der Importeur Herstellerpflichten übernimmt. Ich formuliere das gleich eindeutiger.
Besten Dank und viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
ich würde gerne wissen, was man sich unter der Verantwortlichkeit:
„Konformität der Medizinprodukte wird in Übereinstimmung mit dem QM-System (vor der deren Auslieferung) geprüft (Artikel 10(9))“
vorstellen kann. Was genau ist hier verlangt, ist die Verantwortliche Person etwa für die Chargenfreigabe zuständig?
Sonst fällt mir dazu nichts ein, vlt. haben Sie an dieser Stelle mehr Informationen.
Mit freundlichen Grüßen, Konstantin Frank
Sehr geehrter Herr Frank,
die Konformität bezieht sich nicht nur auf die Produktion, sondern auch auf das Design. Was die verantwortliche Person machen kann, ist zu überprüfen, ob eigen Vorgaben zur Entwicklung und Produktion angemessen sind und auch eingehalten werden. D.h. sie kann prüfen, ob das QM-System funktioniert. Ähnliche steht das auch im MDCG Dokument. Eine Chargenfreigabe kann aber muss nicht zur Aufgabe der PRRC zählen.
Ich erwarte, dass es künftig weitere „Guidance“ zu diesem Thema gibt.
Beste Grüße
Christian Johner