
Medizinproduktehersteller, aber auch Hersteller in allen anderen Lifescience-Bereichen wie der Pharmaindustrie, sind zu einem regelmäßigen „Regulatory Update“ verpflichtet. Denn so eigenartig es klingt: Regulatorische Anforderungen verpflichten Hersteller dazu, fortlaufend die Änderungen der regulatorischen Anforderungen zu überwachen, zu bewerten und notwendige Maßnahmen zu treffen.
Bei Tausenden dieser regulatorischen Anforderungen den Überblick zu behalten, ist eine Herausforderung. Hersteller sollten genau verstehen, welche typischen Fehler es zu vermeiden gilt, um einerseits Sicherheit bei Audits zu erlangen und andererseits unnötige Aufwände für ihr „Regulatory Update“ zu ersparen.
1. Regularien, die ein „Regulatory Update“ verfolgen muss
Die Landkarte der Regularien ist ebenso komplex wie umfangreich:
- Nationale Gesetze wie MPG, MPDG, Heilmittelgesetz, Arzneimittelgesetz (AMG), Food, Drug & Cosmetic Act
- Nationale Verordnungen wie MPBetreibV, MPAIMV
- Weitere Publikationen der Nationalstaaten wie des NAKI, des PEI oder des RKI
- EU-Verordnungen wie MDR und IVDR, Arzneimittelverordnung
- EU-Leitlinien wie die MDCG-Dokumente oder EMA-Guidances
- Hunderte nationale und internationale Normen wie ISO 13485 und ISO 14971
- Common Specifications
- Weit über 600 FDA Guidance Documents, z. B. zur Cybersecurity und zur Vigilanz
- Implementierungsleitfäden, z. B. zur IT-Security und zum Machine Learning
- Veröffentlichungen von Organisationen wie IMDRF und NB-Med
Eine weitere Übersicht zu Regularien mit den jeweiligen Links finden Sie auf unserer Seite Regulatory Affairs.
2. Regulatorische Anforderungen an die Recherche regulatorischer Anforderungen
a) EU-Verordnungen (MDR, IVDR)
Hersteller müssen aktuelle Normen und Common Specifications kennen und berücksichtigen
Medizinprodukte müssen laut EU-Verordnungen den grundlegenden Sicherheits- und Leistungsanforderungen des Anhangs I entsprechen und dem „allgemein anerkannten Stand der Technik“ genügen. Hersteller sind verpflichtet, bei der Konformitätsbewertung die angewendeten harmonisierten Normen, Common Specifications oder „sonstigen Lösungen“ anzugeben.
Gemäß Artikel 10 dürfen Hersteller harmonisierte Normen nutzen, um den Stand der Technik nachzuweisen, und müssen diese entsprechend verfolgen. Die MDR fordert ausdrücklich:
„Änderungen der harmonisierten Normen oder der GS, auf die bei Erklärung der Konformität eines Produkts verwiesen wird, werden zeitgerecht angemessen berücksichtigt.“
MDR, Artikel 10
Fazit: Medizinproduktehersteller müssen Normen und grundlegende Spezifikationen (GS, Common Specifications) aktiv nachverfolgen, um den Stand der Technik zu kennen und nachzuweisen.
Lesen Sie hier mehr zum Thema harmonisierte Normen und Stand der Technik.
Da die Harmonisierung von Normen ins Stocken gekommen ist, erwarten die Benannten Stellen i. d. R., dass die Hersteller die neuesten Versionen der Normen berücksichtigen.
Hersteller müssen im QM-System festlegen, wie sie rechtliche Anforderungen nachverfolgen.
MDR und IVDR haben die Anforderungen an QM-Systeme deutlich erhöht. So fordert die MDR:
Diese Verfahren und Techniken haben speziell Folgendes zum Gegenstand:
MDR. Anhang IX, Abschnitt 2.2
— das Konzept zur Einhaltung der Regulierungsvorschriften, einschließlich der Prozesse zur Feststellung der einschlägigen rechtlichen Anforderungen, Qualifizierung, Klassifizierung, Handhabung von Gleichartigkeit, Wahl und Einhaltung der Konformitätsbewertungsverfahren
b) ISO 13485
„Regulatory Update“ als explizit geforderte Tätigkeit innerhalb des QM-Systems
Auch die ISO 13485 adressiert explizit das Thema. Sie schreibt:
„Die oberste Leitung muss sicherstellen, dass die Kundenanforderungen und anwendbare regulatorische Anforderungen ermittelt und erfüllt werden.“
DIN EN ISO 13485:2016, Kapitel 5.2
Wie wichtig diese Überwachung ist, macht Kapitel 5.6 der Norm (Managementbewertung) klar. Die Managementbewertung muss „anwendbare neue oder überarbeitete regulatorische Anforderungen“ als Input bewerten.
Als Ergebnis dieser Bewertung muss das Management „Änderungen [festlegen], die erforderlich sind, um auf anwendbare neue oder überarbeitete regulatorische Anforderungen zu reagieren“.
Produktspezifische regulatorische Anforderungen
Zusätzlich fordert die ISO 13485, dass die Hersteller pro Produkt die Stakeholder-Anforderungen ermitteln. Damit sind die Anforderungen der Kunden gemeint, aber auch die regulatorischen:
„Die Organisation muss Folgendes ermitteln: […] anwendbare regulatorische Anforderungen bezüglich des Produkts;“
ISO 13485 Abschnitt 7.2.1
Doch mit der Ermittlung dieser Anforderungen ist es nicht getan:
„Die Organisation muss die Anforderungen bezüglich des Produkts bewerten. Diese Bewertung muss […] sicherstellen […], dass die anwendbaren regulatorischen Anforderungen erfüllt sind; [und], dass die Organisation in der Lage ist, die festgelegten Anforderungen zu erfüllen.“
ISO 13485 Abschnitt 7.2.2
c) ISO 20416 „Medical devices – Post-market surveillance for manufacturers“
Die ISO 20416 betrachtet das „Regulatory Update“ als Teil der Post-Market Surveillance. Die Norm fordert explizit:
„Medical device organizations should monitor applicable regulatory requirements for any change to evaluate upcoming gaps, and plan for continued compliance. Standards, guidances and best practices are typically not mandatory requirements (see regulatory requirements) but describe the state of the art.
ISO 20416 (Draft)
Changes in regulatory requirements, standards, guidances and best practices can suggest a change in the state of the art, impacting design and development inputs and potentially requiring design and development changes.”
d) 21 CFR part 820
Die FDA formuliert nicht explizit, dass die Hersteller die regulatorischen Anforderungen und Normen zu ermitteln haben. Allerdings fordert sie:
“Where process controls are needed they shall include:
21 CFR § 820.70
[…]
Compliance with specified reference standards or codes;“
Dass FDA-Inspektoren davon ausgehen, dass die Hersteller alle Regularien kennen und befolgen, dürfte jedoch selbstverständlich sein.
3. Hürden und Herausforderungen
Die Erkenntnis, dass Hersteller die regulatorischen Anforderungen kennen und befolgen müssen, ist banal. Weniger banal sind die Herausforderungen, die sie dabei zu bewältigen haben:
- Anwendbare regulatorische Anforderungen tatsächlich finden
Die erste Hürde, die Hersteller bewältigen müssen, besteht darin, die relevanten regulatorischen Anforderungen zu ermitteln. Je größer die Anzahl der Länder, in denen ein Produkt vermarktet werden soll, desto länger wird die Liste dieser Regularien sein. - Aufwand für die Überwachung und Bewertung
Wenn ein Hersteller alle anwendbaren Regularien gefunden hat, wird ihn deren schiere Menge erschlagen: Mehrere Hundert anwendbare Regularien sind nicht ungewöhnlich für einen Hersteller.
Jedes Dokument bedeutet einen zusätzlichen Aufwand für das- Überwachen,
- Lesen und Verstehen,
- Erkennen der Unterschiede,
- Ableiten der notwendigen Konsequenzen. Diese können bis zur Produktänderung oder im unwahrscheinlichen Extremfall gar einem „Recall“ reichen.
- Heterogenität der Informationsquellen
Doch wie bleibt man auf dem Laufenden? Jede Quelle nutzt andere Medien, um Änderungen zu kommunizieren: Newsletter, RSS-Feeds und Twitter-Nachrichten sind Beispiele. Leider fehlen oft Informationen, d. h. man ist gezwungen, aktiv nach Änderungen zu suchen. - Anforderungen nur in der Landessprache
Doch selbst, wenn man erfährt, dass es neue Regularien gibt, ist das Problem nicht gelöst: Viele Länder und Behörden wie z. B. die chinesische NMPA publizieren einen Teil der Regularien nicht oder nur verzögert auf Englisch bzw. Deutsch. - Verständlichkeit
Dass ein Regularium auf Deutsch oder Englisch vorliegt, gewährleistet noch nicht, dass man die darin formulierten Anforderungen auch versteht. Der Artikel 120(3) der MDR ist ein unrühmliches Beispiel für mangelnde Verständlichkeit. - Kosten
Eine weitere Hürde – insbesondere für Startups – bilden die Kosten. Beispielsweise verlangen Organisationen wie DIN, ISO oder IEC häufig mehrere Hundert Euro pro Norm. In Summe sind mehrere Tausend Euro pro Jahr keine Ausnahme. - Mit den Ergebnissen umgehen
Der Prozess ist an dieser Stelle noch lange nicht abgeschlossen. Das Monitoring ist nur der erste Schritt. Nun muss sichergestellt werden, dass die Informationen an die richtigen Rollen weitergeleitet sowie entsprechende Maßnahmen zur Umsetzung in einem vorgegebenen oder mindestens angemessenen Zeitrahmen ergriffen werden.
4. Typische Fehler beim „Regulatory Update“
Das Johner Institut beobachtet regelmäßig die folgenden Fehler, die bei Audits oder bei der Zulassung zu unliebsamen Überraschungen führen:
- Die Hersteller haben nicht alle Anforderungen ermittelt. Insbesondere fehlen häufig nationale Vorgaben und Leitlinien, die zwar nicht verbindlich sind, aber dennoch gefordert werden.
Manchmal sind die Regularien zwar im Unternehmen bekannt, allerdings pflegt jede Abteilung eigene Listen. Diese stehen dann beim Audit und beim Management-Review nicht in konsolidierter Version zur Verfügung. - Die Ermittlung der Änderungen erfolgt zu spät bzw. zu selten. Es ist peinlich, wenn man im Audit feststellt, dass die Änderung einer Leitlinie, die vor einem halben Jahr publiziert wurde, nicht bekannt ist.
- Meist ist der Grund dafür darin zu finden, dass es keinen Prozess gibt oder Verantwortlichkeiten nicht klar geregelt sind. Der Regulatory Affairs Manager hat sich auf die Ländergesellschaft verlassen, der Produktmanager auf Regulatory Affairs.
- Doch die Kenntnis der Änderungen reicht nicht aus. Regelmäßig fehlt eine angemessene Analyse, worin die Änderungen bestehen, sowie eine Bewertung, was diese bedeuten. Solch eine Bewertung bedarf meistens des Risikomanagements, das aber nicht einbezogen wurde.
- Mangelnde Prozesskontrolle bei der Umsetzung: In den meisten Unternehmen werden die Informationen per E-Mail oder Chatfunktionen verteilt, was häufig dazu führt, dass die Informationen nicht bei den richtigen Rollen ankommen. Der aktuelle Status der Umsetzung ist nicht transparent einsehbar. Im schlechtesten Fall arbeiten mehrere Personen unabgestimmt an Lösungen, die nicht zueinander passen.
- Unvollständiges Management-Review: Die Führung prüft nur, ob nach neuen Regularien gesucht wurde. Sie bewertet aber weder die Güte der Überwachung noch Güte und Umsetzung der Maßnahmen, die sich aus neuen oder geänderten Regularien ergeben.
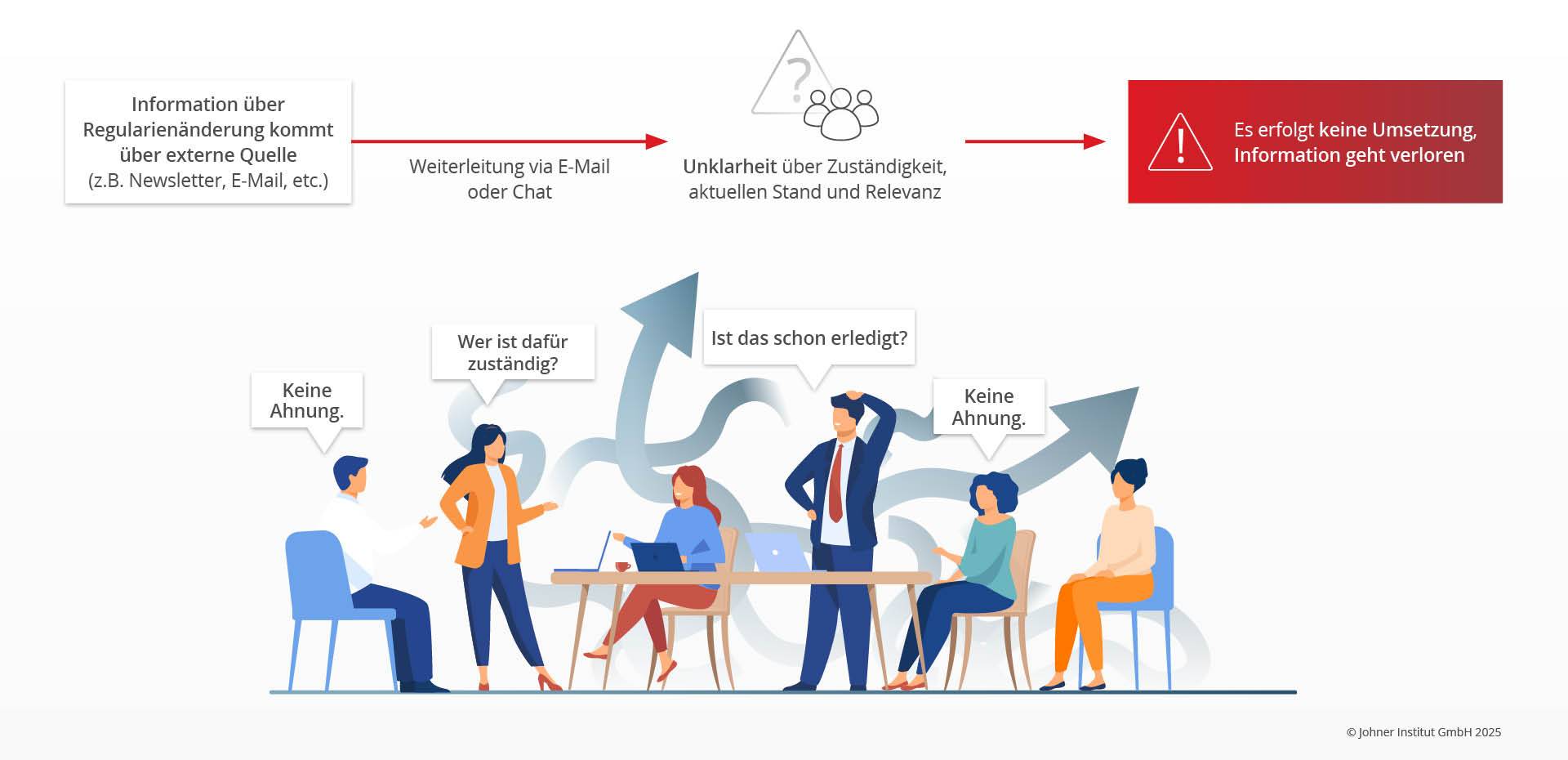
5. „Regulatory Update“: Best Practices
a) Prozess festlegen
Unabhängig davon, ob ein Prozess bzw. ein Verfahren für das „Regulatory Update“ gefordert wird: Legen Sie solch einen Prozess bzw. solch ein Verfahren fest. Eine entsprechende Verfahrensanweisung könnte die folgenden Schritte umfassen:
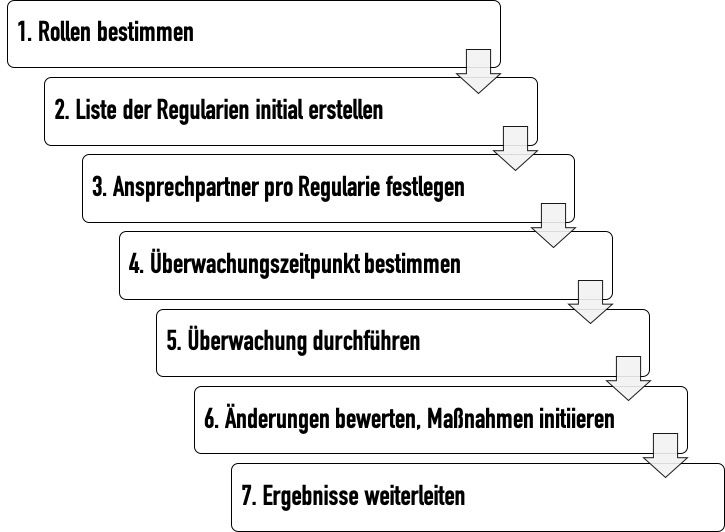
- Schritt: Verantwortliche Rollen bestimmen
Legen Sie die Rollen fest, die verantwortlich sind, die Liste der regulatorischen Anforderungen initial zu erstellen und fortlaufend zu überwachen. Zu den typischen Rollen zählen Produktmanager, Entwickler, Regulatory-Affairs- und Qualitätsmanager, die Rechtsabteilung sowie Landesgesellschaften. - Schritt: Liste der regulatorischen Anforderungen initial erstellen
Lassen Sie von den zugewiesenen Rolleninhabern nun produktspezifische und unternehmensspezifische Listen der anwendbaren Regularien erstellen. Üblicherweise entstehen mehrere Listen, die es dann zu konsolidieren gilt. - Schritt: Ansprechpartner pro Regularium festlegen
Legen Sie jeweils die Personen fest, die Änderungen an den Regularien initial und detailliert bewerten und als kompetente Ansprechpartner dienen. - Schritt: Überprüfungszeitpunkte bestimmen
Bestimmen Sie den Zeitpunkt, an dem die Regularien überwacht werden.- Das kann im Turnus erfolgen und beispielsweise einen Monat vor dem Management-Review stattfinden. Ein jährlicher Turnus ist nicht ausreichend. Wir empfehlen mindestens ein vierteljährliches Review, im besten Fall monatlich.
- Die Überwachung kann auch an Lebenszyklusphasen gekoppelt sein (z. B. zu Beginn und Ende der Entwicklung oder bei Design Reviews).
- Die Überprüfung kann auch ereignisbasiert erfolgen.
- Schritt: Überwachung durchführen
Anhand dieser Vorgaben überwachen die gewählten Rollen bzw. Personen die identifizierten Regularien in der festgelegten Frequenz und dokumentieren, ob es Änderungen gab. - Schritt: Änderungen bewerten und mögliche Maßnahmen initiieren
Falls es Änderungen gibt, informieren die Rollen aus dem 5. Schritt die Ansprechpartner und bitten,- diese Änderungen zu analysieren,
- die Relevanz der Änderung zu bewerten (hierfür Template verwenden und ggf. Risikomanager einbeziehen),
- notwendige Maßnahmen anzustoßen (Abzweigung in andere Prozesse wie Entwicklungsprozess, Wartungsprozess, Problemlösungsprozess, Prozesse für Korrekturmaßnahmen und Vorbeugemaßnahmen, Vigilanzprozess) und
- all dies zu dokumentieren.
- Schritt: Kontrolle der Implementierung der Maßnahmen
Die Umsetzung der Maßnahmen muss kontinuierlich nachverfolgt werden, um zu vermeiden, dass z. B. durch Behörden vorgegebene Umsetzungsfristen nicht überschritten werden. - Schritt: Ergebnisse weiterleiten
Unabhängig davon, ob Änderungen gefunden wurden, müssen die Ergebnisse als Input für andere Prozesse dienen und an diese weitergeleitet werden. Dazu zählen insbesondere Prozesse wie das Management-Review und die Post-Market Surveillance.
b) Prozess automatisieren oder automatisierten Prozess nutzen
Wie herausfordernd die Tätigkeit des „Regulatory Update“ ist und welche typischen Fehler den Herstellern dabei unterlaufen, haben die Abschnitte 3 und 4 bereits beleuchtet.
Um diese Fehler sowie Aufwände für repetitive Tätigkeiten zu vermeiden, sollten Hersteller in Betracht ziehen, diese Tätigkeiten zu automatisieren. Computer erledigen dies schnell, fehlerfrei und kostengünstig. Dedizierte Dokumentations- und Task-Management-Tools unterstützen die Prozesskontrolle und stellen sicher, dass alle Informationen transparent an einem Ort verfügbar sind und keine Fristen übersehen werden.
Das Johner Institut hat beispielsweise Crawler implementiert, die kontinuierlich Webseiten und Datenbanken überwachen und die gefundenen Änderungen einem Überwachungsteam zur Bewertung vorlegen. Ein auf die Anforderungen von Herstellern abgestimmter Workflow leitet kontrolliert und transparent durch den Prozess vom Monitoring bis zur Implementierung der neuen regulatorischen Anforderungen.
Übrigens: Dieses Team übernimmt die Überwachung der Regularien für viele Hersteller. Dadurch ergeben sich weitere Skalierungseffekte, von denen die Hersteller profitieren.
Durch unsere Arbeit mit vielen Herstellern konnten wir einen klassischen Prozess in vier Schritten ableiten, durch den sie im Regulatory Radar strukturiert geleitet werden:
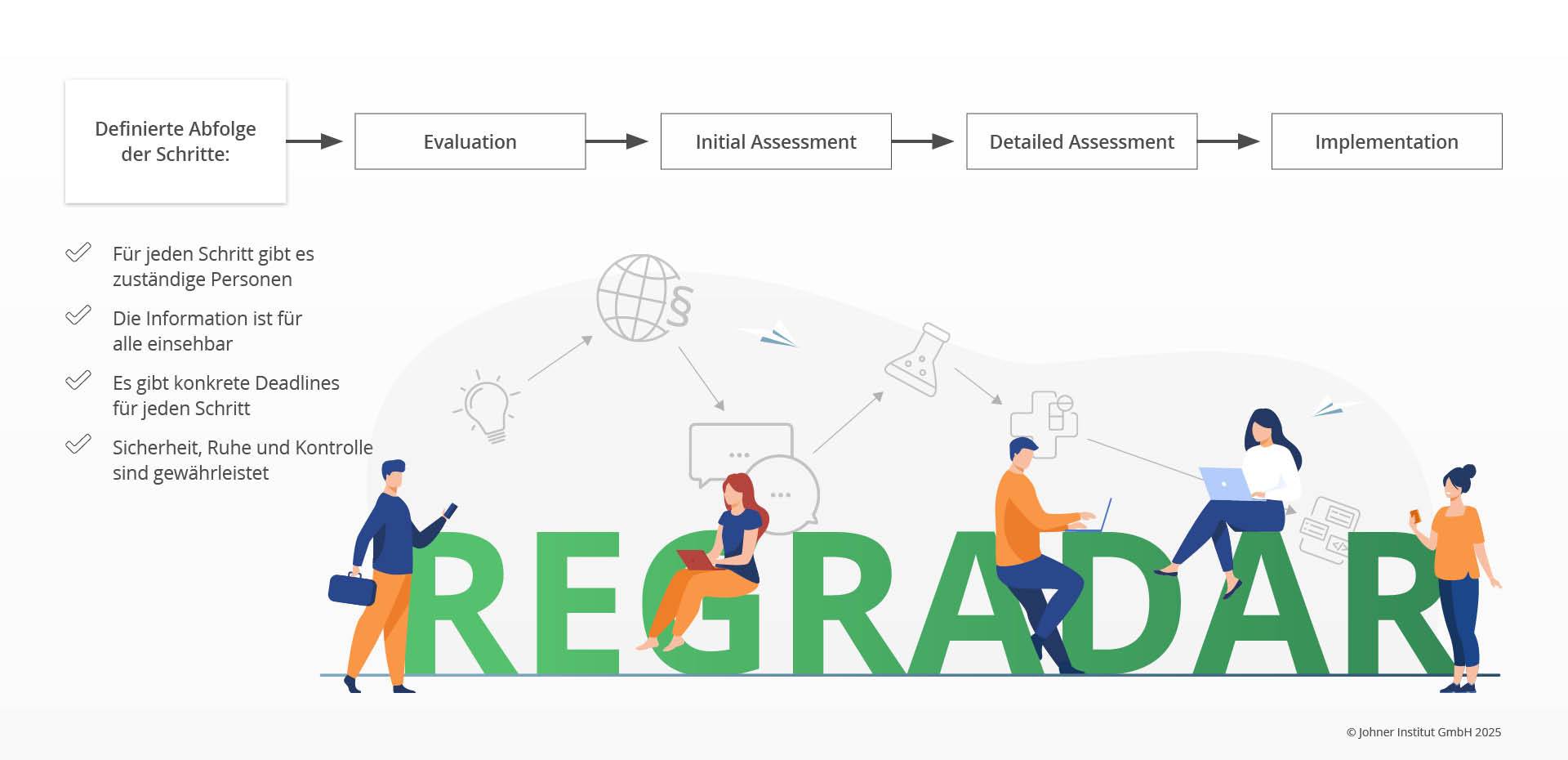
Folgende Aktivitäten sind Teil der einzelnen Schritte:
- Evaluation: Erste Einschätzung, ob die Änderung relevant sein könnte
- Initial Assessment: Etwas detailliertere Einschätzung, welche Auswirkungen die Änderung haben könnte
Typische Fragen im Initial Assessment können sein:- Ist die Einhaltung der Regularie zwingend erforderlich oder „nur“ empfohlen?
- Welchen geografischen Impact hat die Änderung (global oder lokal)?
- Welche Prozesse können davon betroffen sein?
- Welche Produkte aus dem Portfolio können betroffen sein?
- Detailed Assessment: Festlegung der Maßnahmen zur Umsetzung, meist durch Subject Matter Experts
- Implementierung: Umsetzung und Dokumentation der Maßnahmen
Hersteller, die nicht einen automatisierten Service wie den Regulatory Radar des Johner Instituts nutzen, sondern die Überwachung selbst automatisieren wollen, sollten auf Folgendes achten:
- Die Investition rechnet sich umso mehr, je mehr Regularien zu überwachen sind und je häufiger diese Überprüfung stattfindet.
- Es bedarf einer komplexen Geschäftslogik, mit der „false positives“ und „false negatives“ bestmöglich vermieden werden. Manche Webseiten ändern beispielsweise täglich die IDs im HTML-Code, um Crawler abzuschrecken. Die technische Qualität mancher Seiten ist bedenklich.
- Eine fortlaufende Anpassung der Software ist notwendig, weil sich die Struktur der verfügbaren Informationen insbesondere auf Webseiten regelmäßig ändert.
- Die Software unterliegt den Anforderungen der ISO 13485 und ist zu validieren bzw. bei Anpassungen zu revalidieren (siehe Computerized Systems Validation).
- Der Prozess ist mit dem Monitoring keineswegs beendet. Die Ergebnisse müssen konsistent und transparent weiterverarbeitet werden. Hierfür bedarf es evtl. weiterer Lösungen mit entsprechenden Herausforderungen.
c) Prozess überwachen durch KPIs
Wie auch bei anderen Prozessen ist es sinnvoll, das Funktionieren des Prozesses durch Kennzahlen (Key Performance Indicators, KPIs) zu überwachen. Welche Kennzahlen sind für diesen Prozess relevant? Hier einige Beispiele:
- Anzahl der offen/nicht bearbeiteten Änderungsmeldungen (gibt es evtl. Kapazitätsengpässe im Regulatory Affairs Team?)
- Anzahl der überfälligen Maßnahmen (hier drohen Nicht-Konformitäten)
- Anzahl der Bearbeitungen, die sich im Review befinden (gibt es evtl. Kapazitätsengpässe im Freigabeprozess?)
- Zeitraum von der Erfassung einer Änderung bis zur Umsetzung der festgelegten Maßnahmen (sind Trends erkennbar?)
- Anzahl der abgelehnten Reviews der Bewertungen (kann bspw. auf Wissensengpässe hindeuten)
Im Fall der Nutzung eines automatisierten Systems sollten diese Kennzahlen natürlich direkt vom System mitgeliefert werden. Das erspart Zeit und Aufwand bei der Kennzahlerhebung. Durch Alarme, Benachrichtigungen etc. werden verantwortliche Mitarbeiter direkt auf mögliche Probleme und somit evtl. entstehende Nichtkonformitäten hingewiesen.
6. Zusammenfassung
Hersteller müssen die Änderungen der regulatorischen Anforderungen proaktiv überwachen und bewerten. Das ist selbst eine regulatorische Anforderung.
Zwar sind Hersteller nicht explizit verpflichtet, ein Verfahren dafür festzulegen; dies zu tun, ist aber sehr zu empfehlen. Die MDR fordert zumindest ein „Konzept“.
Die wachsende Anzahl der Regularien und die Frequenz der Änderungen bedeuten für die Hersteller hohe Aufwände für die Überwachung und Bewertung. Daher empfiehlt es sich, diese Tätigkeiten bestmöglich zu automatisieren oder outzusourcen, wie das beispielsweise die Pharmabranche längst macht.
Wer den Überwachungsprozess automatisiert, muss die Systeme validieren.
Hersteller dürfen sich bei der Überwachung keineswegs auf regulatorische Anforderungen speziell für Medizinprodukte beschränken. Vielmehr müssen sie auch Regularien z. B. zum Datenschutz, zur Produktsicherheit, zum Abfallmanagement oder aus dem Sozialrecht recherchieren und befolgen.
Zudem sollten Hersteller das „Regulatory Update“ nicht als isolierten Prozess sehen, sondern als Teil der Post-Market Surveillance und der Regulatory Intelligence des Unternehmens.
Erfahren Sie hier mehr über den Regulatory Radar.
Änderungshistorie
- 2025-04-02: Artikel weitestgehend überarbeitet
- 2020-04-28: Artikel initial erstellt



Danke für diesen Artikel.
Wir nutzen auch den Regulatory Radar von Johner und haben eine deutlich bessere Übersicht als früher, als wir dies manuell gemacht haben und vermutlich auch zu wenige Quellen überprüft haben.
Ich habe eine Frage zum Prozess und den nachgeschalteten Prozessen, worauf im Text auch kurz eingegangen wird.
Die regulatorischen Änderungen müssen bewertet werden, ob diese anwendbar sind auf das eigene QMS bzw. auf die Produkte. Wenn dies der Fall ist, sind hierzu Maßnahmen notwendig, um die Prozesse entsprechend anzupassen.
Handelt sich bei diesen Maßnahmen grundsätzlich um Vorbeugungsmaßnahmen nach ISO 13485 8.5.3 inkl. Verifizierung und Wirksamkeitsprüfung, da eine vermeintliche Nichtkonformität gegenüber der Regularie verhindert werden soll?
Ist ein einfacherer Weg möglich, da dies bei den ständigen Änderungen der Regularien den CAPA-Prozess stark belasten würde?
Lieber Herr Handt,
meiner Meinung nach ist das nicht bei jeder Änderung notwendig. Es hängt stark davon ab, ob es eine Regularie ist, die zwingend anzuwenden ist und auch über die Art der Änderung. Beispielsweise können Präzisiserungen von Vorgaben oder auch formale Änderungen zwar relevant sein, aber führen nicht zwangsläufig zu Nichtkonformitäten. Ich rate hier zu einer Fall-zu-Fall-Entscheidung, ob der CAPA-Prozess anwendbar ist oder nicht.
Beste Grüße,
Andrea Seeck
Guten Tag,
Vielen Dank für den Artikel.
Ich glaube einen Zitierfehler gefunden zu haben.
„Änderungen der harmonisierten Normen oder der GS, auf die bei Erklärung der Konformität eines Produkts verwiesen wird, werden zeitgerecht angemessen berücksichtigt.“ Dieses Zitat stammt aus Artikel 10 und nicht 8.
Viele Grüße
Vielen lieben Dank für den Hinweis Herr Schwanau. Das stimmt natürlich und ich habe es direkt korrigiert.
Beste Grüße,
Andrea Seeck