Medizinproduktehersteller haben hohe Erwartungen an Regulatory Information Management Systems (RIMS). Kosten und Aufwände dafür sind immens und meist viel höher als geschätzt. Der Nutzen steht hingegen nicht fest.
Dieser Artikel liefert Ihnen die entscheidenden Hinweise,
- ob Sie ein Regulatory Information Management System benötigen und
- worauf Sie bei dessen Auswahl und Einführung achten sollten.
1. Definition: RIMS
Ein Regulatory Information Management System (RIMS) ist ein Software-System, das Pharma- und Medizinproduktehersteller dabei unterstützt, regulatorische Informationen zu erfassen und zu verwalten, um regulatorische Konformität sicherstellen zu können. Es handelt sich um eine zentrale Plattform, die die Erfassung, Verwaltung und Bereitstellung von regulatorischen Daten und Dokumenten ermöglicht.
Quelle: Johner Institut
2. Ziele von RIMS
Vom Einsatz eines Regulatory Information Management Systems versprechen sich Pharma- und Medizinproduktehersteller mehrere Vorteile:
- Höhere Compliance, minimierte regulatorische Risiken
Ein RIMS soll dabei helfen, Unterlagen auf Vollständigkeit zu prüfen, und an überfällige Aufgaben und Fristen erinnern. Oft bieten RIMS auch eine Übersicht über relevante regulatorische Anforderungen.
Zudem verfolgen einige RIMS regulatorische Änderungen und informieren darüber, sodass die Hersteller durch entsprechende Maßnahmen die Konformität fortlaufend gewährleisten können. - Verbesserte Effizienz, beschleunigte Zulassung
Das RIMS soll die Zusammenarbeit innerhalb des Unternehmens und mit den Zulassungsstellen vereinfachen. Dazu regelt es klar die Abläufe und automatisiert teilweise die Aufgaben.
Die Systeme erleichtern die Suche nach Informationen und erlauben allen Beteiligten – von der Entwicklung bis zum Post-Market-Team – gemeinsam an Daten zu arbeiten.
Die Entwicklungsabteilung muss von Beginn an wissen, was regulatorisch gefordert ist. Die Regulatory Affairs Abteilung muss von Beginn an wissen, was entwickelt werden soll, um bei der regulatorischen Strategie zu helfen.
Andernfalls droht die Gefahr, dass Produkte nicht, nicht mit den geplanten Features oder nicht in der geplanten Zeit auf den Markt kommen.
- Verbesserte Datenqualität
Die Daten liegen zentral vor und können zentral eingesehen werden. Beides trägt dazu bei, dass fehlerhafte Daten vermieden oder zumindest schnell bereinigt werden. Beides wiederum ist notwendig, um Entscheidungen auf der Basis von verlässlichen Informationen zu treffen. - Verlässliche Informationen als Entscheidungsgrundlage
Die gemeinsame Plattform und zentrale Aufbereitung der Daten verschafft den Unternehmen Übersicht, z. B. über den Fortschritt von Zulassungsprojekten.
Diese Transparenz ist für fundierte Entscheidungen notwendig, z. B. die Festlegung des Beginns einer Kampagne zur Markteinführung.
3. Funktionen eines RIMS
Um die eben genannten Ziele zu erreichen, bieten RIMS zahlreiche Funktionen an.
a) Verwaltung regulatorischer Anforderungen
RIMS unterstützen den Umgang mit den regulatorischen Anforderungen:
- Zentrale Speicherung von regulatorischen Dokumenten, Richtlinien und Standards
- Verwaltung von regulatorischen Änderungen und Aktualisierungen
- Nachverfolgung notwendiger Maßnahmen
b) Management der Zulassungsverfahren
- Verfolgung der Zulassungsverfahren
- Unterstützung bei der Einreichung von regulatorischen Dokumenten und Anträgen
- Verwaltung von Technischen Dokumentationen
- Überwachung von Compliance-Fristen und -Aufgaben
c) Post-Market Surveillance
- Automatisiertes Sammeln und Filtern der Daten im Rahmen der Post-Market Surveillance
- Analyse und Trendberechnung dieser Daten
- Unterstützung bei der Berichtserstellung (z. B. PSUR)
- Nachverfolgung notwendiger Maßnahmen
d) Sonstiges
- Berichterstattung und Analysen im Bereich der regulatorischen Informationen
- Integration in andere Systeme (siehe Kapitel 3)
4. Einbettung eines RIMS in die Systemlandschaft
a) Übersicht über die Systeme
Um einen möglichst durchgängigen Informationsfluss zu erlauben, müssen Regulatory Management Systeme mit anderen Informationssystemen zusammenarbeiten.
- Dokumentenmanagement-Systeme (DMS)
Besonders relevant ist die Integration eines RIMS in ein DMS. Nur so wird die rechtssichere Speicherung, Verwaltung und Versionierung von regulatorischen Dokumenten sichergestellt.
Die Dokumente in ihren jeweiligen Versionen müssen den Zulassungsverfahren eindeutig zugeordnet werden können. Es ist auch notwendig, jederzeit eine Übersicht zu haben, wann welche Dokumente in welcher Version nachgereicht wurden. - ALM- und PLM-Systeme
In den Systemen für Application Lifecycle Management und Product Lifecycle Management führen die Hersteller wichtig Daten wie die Inhalte eines „Design History Files“, die teilweise für die Einreichung notwendig sind. - ERP-Systeme
Systeme für Enterprise Resource Planning (ERP) speichern relevante Produktstammdaten. Bei vielen Herstellern erfolgt die Zuordnung von Komponenten und Bauteilen zu den einzelnen Medizinprodukten ebenfalls im ERP-System.
Diese Daten sind für Zulassungsverfahren relevant und sollten daher nahtlos in das RIMS übernommen werden können. Die Chargeninformationen sind für die Rückverfolgbarkeit von Produkten und Bauteilen und für Rückrufe sowie für die Einhaltung der Anforderungen an die Lieferkette unerlässlich. - Qualitätsmanagementsysteme (QMS)
Hersteller verwalten im QMS u. a. Vorgabedokumente und verweisen auf Aufzeichnungen wie die Ergebnisse der Verifizierungs- und Validierungsaktivitäten. Diese Nachweise müssen Hersteller bei der Zulassung zur Verfügung stellen. Dies bedingt eine Integration des RIMS in das QMS bzw. das System, das die Nachweise lenkt. - Systeme der Behörden und Benannten Stellen
Behörden und Benannte Stellen verlangen zunehmend, die Zulassungsunterlagen in digitaler Form einzureichen. Dazu bieten sie Schnittstellen an, welche ein RIMS unterstützen sollte.
Es gibt noch weitere Systeme wie CAD-Systeme und Produktinformationsmanagement-Systeme (PIM). Letztere verwalten Produktinformationen wie Gebrauchsanweisungen, Kennzeichnungen und sonstiges Labeling. Auch diese Unterlagen sind Teil der Technischen Dokumentation und damit vom RIMS zu verwalten.
b) Informationsfluss
RIMS bilden somit die Schnittstelle zu den internen und zu den externen Systemen, z. B. bei Behörden und Benannten Stellen (s. Abb. 1).

5. Kriterien zur Auswahl eines RIMS
Üblicherweise nutzen Unternehmen Feature-Listen, um die Anbieter zu vergleichen und das passende Produkt auszuwählen. Solche Kriterienkataloge sind jedoch ungeeignet, um
- herauszufinden, ob ein RIMS überhaupt das richtige System ist,
- die Zukunftsfähigkeit der Auswahl zu beurteilen und
- die Wahrscheinlichkeit abzuschätzen, dass die Einführung erfolgreich sein wird.
Lesen Sie weiter unten „Tipps zur Auswahl und Einführung eines RIMS“.
a) Kriterien, die das Produkt betreffen
Funktionalität
Der erste Impuls besteht darin, zu prüfen, wie vollständig das System die in Kapitel 2 genannten Funktionen anbietet.
Diese Vollständigkeit betrifft Prozesse, beispielsweise:
- Entwicklung
- Zulassung
- Post-Market-Surveillance
- Überwachung regulatorischer Änderungen
Die Vollständigkeit betrifft ebenfalls die Märkte und deren regulatorischen Anforderungen. Ein System aus den USA, welches das Konzept der Benannten Stellen nicht kennt, ist nur bedingt nützlich für einen Hersteller, der seine Produkte in der EU in den Markt bringen will.
Auch sollte das System die Prozesse spezifisch für die eigenen Produkte unterstützen. Agnostische Systeme (wie im Extremfall ein DMS) können die produktspezifischen Inhalte nicht automatisiert prüfen. Diese Arbeit bleibt dann bei den Regulatory Affairs Managern und Qualitätsmanagern hängen.
Ein System kann nur dann prüfen, ob alle wesentlichen Leistungsmerkmale eines Produkts bei den „60601-1-Tests“ berücksichtigt wurden, wenn es das Konzept der wesentlichen Leistungsmerkmale kennt.
Ein System kann nur dann prüfen, ob alle sicherheitsbezogenen Use Scenarios in der Risikomanagementakte vorhanden sind, wenn es diese Entitäten modelliert hat und drauf zugreifen kann. Das ist nicht der Fall bei Systemen, die lediglich PDF und deren Metadaten verwalten.
Datengetriebener statt dokumentengetriebener Ansatz
Ein RIMS sollte einen datengetriebenen und nicht einen dokumentengetriebenen Ansatz verfolgen. Es sind Algorithmen erforderlich, um den erhofften Nutzen zu erreichen:
- Automatisierte Prüfung der Vollständigkeit, Korrektheit und Konsistenz der Daten, z. B. für die Einreichung
- Automatische Analyse der Daten, z. B. für die Post-Market Surveillance
- Dadurch höhere Compliance und kürzere Time to Market
Ein RIMS, das „nur“ den Workflow, die Verwaltung und Versionierung von Dokumenten ermöglicht, ist nicht „fit for future“.
Interoperabilität
In Kapitel 3 ist aufgelistet, mit welchen Systemen ein RIMS zusammenarbeiten muss. Daher ist die Interoperabilität mit diesen Systemen eine Grundvoraussetzung. Andernfalls entsteht ein weiteres Datensilo.
Datensilos haben Redundanzen und damit meist Inkonsistenzen zur Folge. Dadurch erhöhen solche Systeme die regulatorischen Risiken, anstatt sie zu verringern.
Zudem besteht die Gefahr, dass RIMS mit mangelnder Interoperabilität zwar die Regulatory-Affairs-Abteilung lokal optimieren, aber kein globales Optimum erreicht wird in Hinsicht auf z. B. „Compliance“ und Minimierung der „Zulassungsdauer“.
Ideal sind Systeme, welche die Prozesse möglichst „Ende-zu-Ende“ unterstützen. Damit wird die Anzahl der Schnittstellen minimiert und gleichbedeutend die Anzahl der Probleme mit der Interoperabilität (s. Abb. 3).
Hersteller sollten ein Design for registration anstreben. Das bedeutet, dass bereits zu Beginn der Entwicklung die regulatorischen Anforderungen klar sein müssen. Die Entwicklung muss fortlaufend und automatisiert darüber informiert werden, wie vollständig diese Anforderungen bereits erfüllt sind.
Gebrauchstauglichkeit
Ein gebrauchstaugliches Produkt muss zuerst der Zielerreichung (Effektivität), dann der Effizienz dieser Zielerreichung und erst im dritten Schritt der Zufriedenheit der Anwender dienen (s. Abb. 2). Anwender, die ihre Ziele nicht oder nicht effizient erreichen, sind auf Dauer auch nicht zufrieden.
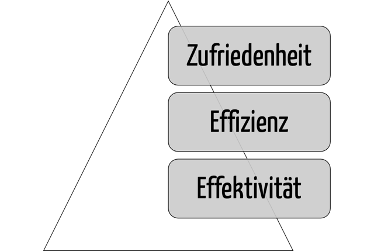
Viele (potenzielle) Anwender verwechseln ein modernes Design mit Gebrauchstauglichkeit. Ein „fancy“ Dashboard ist nicht unbedingt ein User Interface (UI), mit dem die Anwender schnell und ohne unnötige Umwege ihre Aufgaben vollständig und korrekt erfüllen können (s. Abb. 2).
Daher sollten vor der finalen Kaufentscheidung die künftigen Hauptanwender mit dem System gearbeitet haben und maßgeblich in die Entscheidung eingebunden werden.
Nicht-funktionale Anforderungen
Zu den (weiteren) nicht funktionalen Anforderungen zählen:
- Performanz und Skalierbarkeit der Systeme
- Sicherheit und Datenschutz
- Portabilität, d. h. die Fähigkeit, auf den Systemen des (Medizinprodukte-)Herstellers betrieben zu werden. Bei webbasierten Systemen muss der jeweilige Browser der Anwender unterstützt werden.
Auch browserbasierte Systeme benötigen Konnektoren zu den Systemen des Herstellers.
Die ISO 25010 nennt allgemeine Qualitätskriterien für Software.
Kosten
Die Kosten setzen sich meist zusammen aus:
- Lizenzkosten (einmalig, Wartungskosten, Abonnement), abhängig von
- Anzahl der Nutzer (ggf. gestaffelt nach deren Rollen)
- freigeschalteten Modulen / Funktionalitäten
- Nutzungsverhalten, z. B. Anzahl der Produkte oder Einreichungen
- Kosten für den Support
- Kosten für die Einführung (Datenmigration, Parametrierung, Schulung)
- Kosten für den Betrieb der Infrastruktur (falls das System selbst gehostet wird)
- Dauer der Verpflichtung
b) Kriterien, die den Anbieter betreffen
Ein RIMS, das alle Anforderungen zufriedenstellend erfüllt, sollte trotzdem nicht eingeführt werden, wenn der Anbieter nicht den Kriterien genügt.
Klares Zukunftsbild und Roadmap des Anbieters
Die Anbieter müssen eine klare Vorstellung davon haben, wohin sich die regulatorischen Systeme, die Medizinprodukte, die Technologien und sein RIMS entwickeln. Diese Vision des Anbieters sollte präzise formuliert und schriftlich vorliegen. Sie sollte sich mit der eigenen Vorstellung decken.
Der Anbieter sollte klar skizzieren, wie lange er die Einreichung von Technischen Dokumentationen als (PDF-)Dokumente noch als zeitgemäß empfindet und ab wann sein System eine dokumentenfreie Einreichung unterstützen wird.
Einfluss auf die Produktgestaltung
Anbieter von PIMS müssen einen Spagat bewältigen: Einerseits sollten sie möglichst individuell auf die Kundenwünsche eingehen, andererseits müssen sie die konzeptionelle Integrität ihrer Produkte gewährleisten und ihre ökonomischen Interessen vertreten.
Daher sollten Medizinproduktehersteller nur solche Anbieter auswählen, bei denen sie ein „typischer Kunde“ sind. Denn ähnliche Kunden (andere Medizinproduktehersteller) werden ähnliche Anforderungen an den Anbieter stellen, die jener dann einfacher erfüllen kann.
Viele RIMS-Anbieter haben ihre Wurzeln und ihren größten Kundenstamm im Pharmabereich. Daher ist der Einfluss der Medizinproduktehersteller auf die Produktgestaltung meist begrenzt – auch wenn in der Evaluationsphase anderes behauptet wird.
Güte und Verfügbarkeit des Supports
Regulatory Information Management Systems sind komplexe Software-Anwendungen. Daher benötigen sowohl die Anwender als auch Personen, die für die Installation, Konfiguration und den Betrieb verantwortlich sind, einen kompetenten und verfügbaren Support.
Es genügt nicht, dass der Support weiß, wie das eigene Produkt funktioniert. Er muss die regulatorischen Anforderungen verstehen und erklären können, wie diese mit seinem System erfüllt werden können.
Hersteller sollten prüfen,
- in welchen relevanten Zeitzonen der Support
- zu welchen Uhrzeiten verfügbar ist und
- welche Reaktionszeiten er verspricht.
Fachliche Kompetenz des Anbieters
Der rasante technologische Fortschritt insbesondere im Bereich der künstlichen Intelligenz und des Hyper-Scalings hat signifikante Auswirkungen auf die Anbieter von RIMS:
- Viele technischen Herausforderungen lassen sich in naher Zukunft schon einfacher und schneller lösen. Das bedeutet, dass die technischen Kompetenzen bei der Entwicklung von RIMS zwar wichtig bleiben, aber an Bedeutung verlieren.
- Dafür wachsen die Anforderungen an die fachliche Kompetenz des Anbieters. Dieser muss zwingend ein „Subject Matter Expert“ sein. Im Falle der RIMS ist das die Expertise in Regulatorik (bei Medizinprodukten!).
Ein solides Halbwissen genügt nicht, um Systeme mit künstlicher Intelligenz zu trainieren oder deren Ergebnisse fachkundig zu beurteilen.
Die fachliche Expertise ist notwendig, um die Feinheiten der sich ständig ändernden Regulierung zu verstehen und in Algorithmen abzubilden. Ohne eine entsprechende Algorithmik, welche die Prüfung der regulatorischen Anforderungen automatisiert, bleiben RIMS limitiert auf spezialisierte Dokumenten- und Aufgabenverwaltungssysteme.
Damit werden Hersteller nicht in die Lage versetzt, in kürzester Zeit ihre Produkte zuzulassen oder gar in eine kontinuierliche Konformitätsprüfung überzugehen, wie es bei Software künftig notwendig sein wird.
6. Zehn Tipps zur Auswahl und Einführung eines RIMS
Tipp 1: Entscheiden, ob ein RIMS der richtige Ansatz ist
Ein RIMS kann Probleme lösen und auch viele neue Probleme schaffen. Daher sollten Hersteller vor der Einführung zwei Fragen beantworten:
- Ist ein Software-System tatsächlich die Lösung für unsere Probleme?
Die Einführung einer Software wie eines RIMS führt dazu, dass der Hersteller ein weiteres System hat (wenn er dafür kein anderes abschafft). Jedes zusätzliche System verursacht zusätzliche Aufwände und Kosten für Integration, Konfiguration und Betrieb.
Der Nutzen muss diese Aufwände und Kosten rechtfertigen. Regelmäßig gibt es Ursachen für die Probleme (z. B. bezüglich Konformität, Time to Market und Effizienz), die ein RIMS nicht lösen kann.
Falls ein weiteres System eingeführt werden soll: - Ist ein RIMS das geeignete System?
Die meisten RIMS basieren auf konventionellen dokumentenbasierten Abläufe. Damit manifestieren sie diese konventionellen Abläufe und verankern den Hersteller in der (künftigen) Vergangenheit.
Effizienz und Effektivität regulatorischer Prozesse lassen sich nur „Ende-zu-Ende“ optimieren, das heißt in der Entwicklung bis zur Behörde bzw. Benannten Stelle. Lokale Optimierungen (z. B. beim Hersteller) helfen nur bedingt. Daher bedarf es einer durchgängigen Plattform (s. Abb. 3).
Die Ende-zu-Ende-Integration (Entwicklung bis Behörde bzw. Benannte Stelle) ist weiterhin unerlässlich, um eine „Realtime Compliance“ und eine „Continuous Compliance“ zu erreichen. Diese ist insbesondere bei Software-Produkten notwendig, um die immer kürzer werdenden Release-Zyklen zu unterstützen.
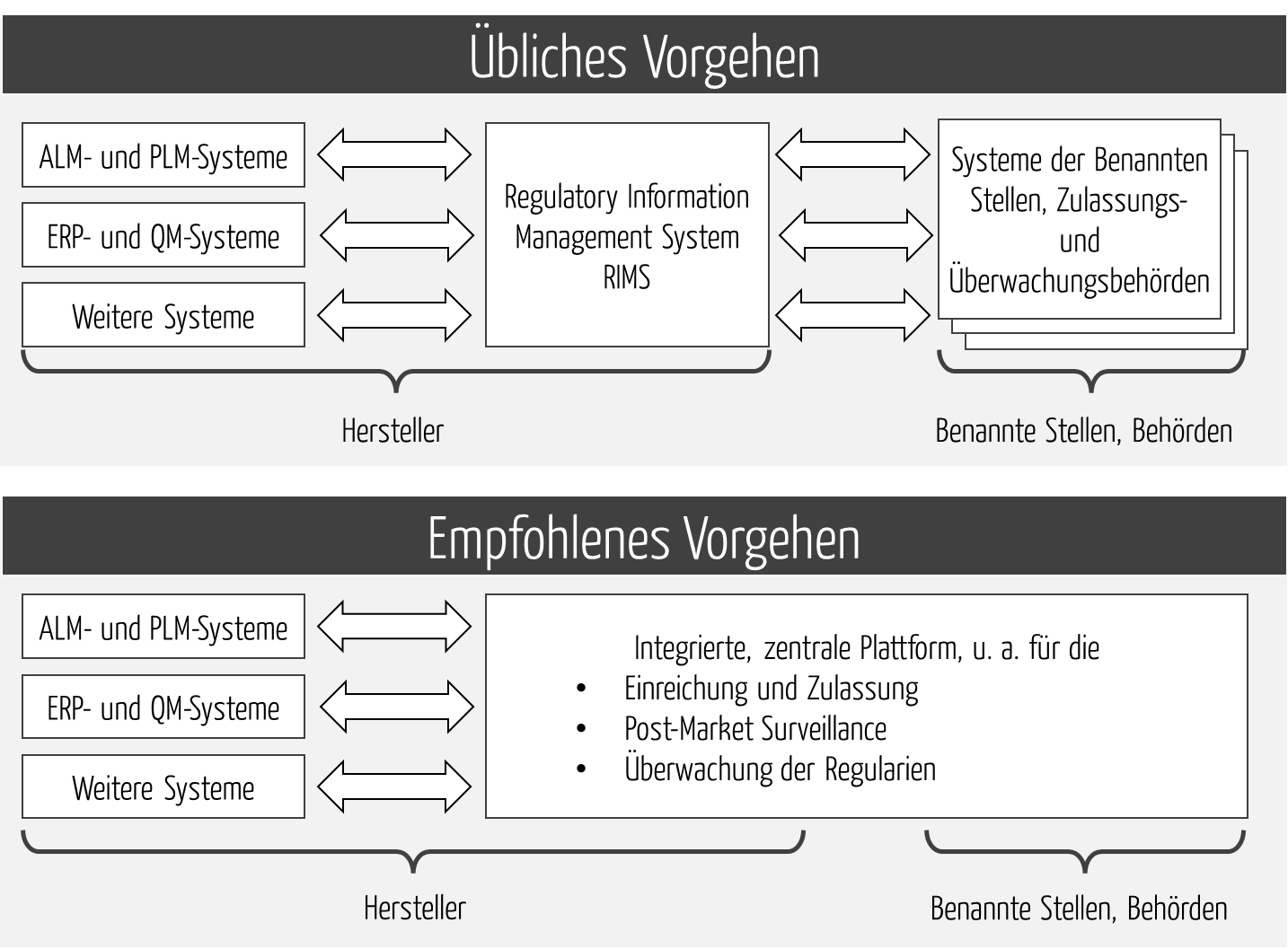
Diese Plattform sollte alle regulatorischen Prozesse unterstützen, nicht nur die Einreichung.
Das wiederum setzt voraus, dass die Plattform nicht nur die internen Systeme integriert, sondern auch die externen (s. Abb. 4).
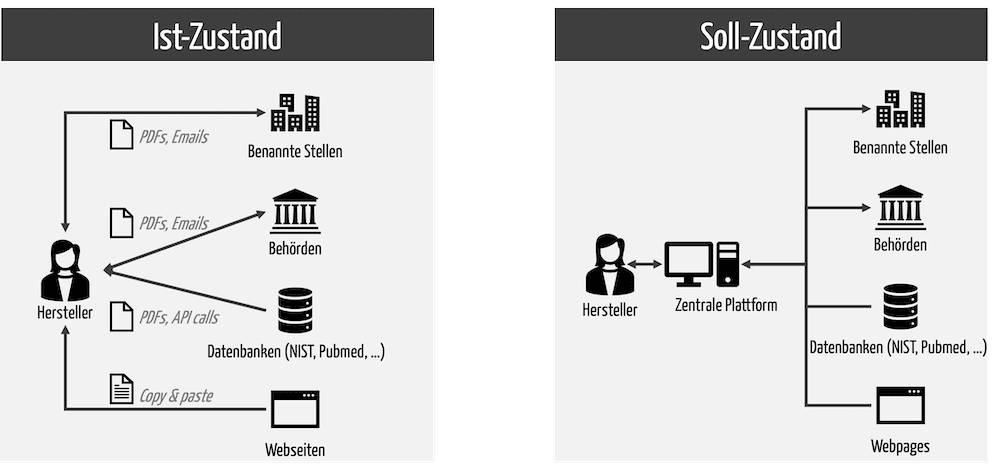
Viele RIMS unterstützen diese umfassende Integration nicht. Das verursacht weitere Schnittstellen und damit Aufwände sowie regulatorische Risiken.
Tipp 2: Nicht von „Feature-itis“ blenden lassen
RIMS-Anbieter beeindrucken mit einer umfangreichen Liste von Features. Aktuell werben viele Anbieter mit der künstlichen Intelligenz ihrer Produkte und preisen diese als Beweis für die Zukunftsfähigkeit ihrer Systeme.
Künstliche Intelligenz sollte aber nur dort zum Einsatz kommen, wo sie tatsächlich Stakeholder-Anforderungen erfüllt, die nicht gleichwertig durch konventionelle Logik erfüllt werden können.
Ein RIMS-Anbieter wirbt damit, dass Technische Dokumentationen schneller mit KI (insbesondere Large Language Models) erstellt werden könnten. Dabei ist die dokumenten-, meist PDF-basierte Einreichung ein Konzept des letzten Jahrtausends.
Es wäre hilfreicher, wenn das System mit konventioneller Logik die Konformität der Inhalte dieser Dokumentation auf Vollständigkeit, Konsistenz und Korrektheit prüft, und die KI nutzt, um externe, unstrukturierte Daten (z. B. klinische Fachliteratur) in interne strukturierte Daten zu überführen.
Die Feature-Listen der Anbieter zielen meist auf die Entscheiderebene beim Hersteller. Das ist nachvollziehbar. Aber die Entscheider sind meist nicht die Personen, die das System später anwenden und betreiben müssen.
Beziehen Sie bei der Auswahl alle Stakeholder ein. Dazu zählt auch Ihre IT-Abteilung, die die Systeme später betreiben muss.
Fragen Sie andere Kunden des Anbieters (Referenzkunden) nach deren persönlichen Erfahrungen. Stellen Sie konkrete Fragen zur Dauer des Projekts, was bei der Einführung geklappt hat und was nicht. Sprechen Sie mit den wirklichen Anwendern und fragen Sie, wie die Daten ins RIMS gelangen und welche Nachbereitung notwendig ist, um die Daten einzureichen.
Tipp 3: Daten aufräumen und strukturieren
Daten sind bei den Herstellern über viele Systeme (siehe oben) und Dateien verteilt. Sie finden sich etwa in ALM-Systemen, in Word- und Excel-Dateien, in E-Mails und Gebrauchsanweisungen. Und manche Daten fehlen ganz.
Die Einführung eines RIMS ändert daran noch nichts. Aber es zwingt die Hersteller, diese Daten „aufzuräumen“:
- Daten vervollständigen
- Redundanzen beseitigen
- Einheitliche Granularität erreichen
- Daten kodieren (z. B. mit IMDRF Codes)
- Referenzen zwischen den Daten herstellen
- Daten korrigieren
- Unnötige Daten eliminieren
Damit das gelingt, benötigen die Hersteller
- ein klares Zielbild dieser Datenstrukturen,
- Datenstrukturen, welche die automatisierte Prüfung der Daten auf Konformität erlauben,
- ein Vorgehensmodell für dieses Aufräumen.
Das Johner Institut unterstützt Hersteller bei dieser Aufgabe. Sie ist die Voraussetzung für die Einführung und den Erfolg eines RIMS oder anderen Systems.
Tipp 4: Vorsicht walten lassen bei Datenmigration
Viele RIMS-Anbieter sehen eine Datenmigration vor. Hersteller sollten sich bewusst sein, dass eine Datenmigration – wie der Name sagt – den Umzug der Daten aus den bisherigen Quellen in das RIMS bedeutet. Solch eine Migration birgt Gefahren:
- Die Daten liegen danach redundant vor.
- Es entstehen Datensilos.
- Im RIMS geänderte Daten fließen nicht zurück in die Primärsysteme wie ALM.
Daher sollten die Hersteller
- sicherstellen, dass es keine Redundanzen gibt,
- festlegen, welches das führende System ist und
- gewährleisten, dass Daten nicht verloren gehen, die für das RIMS unwichtig, aber für andere Zwecke relevant sind.
Tipp 5: Probebetrieb durchführen
Erst bei einem Probebetrieb zeigt sich,
- wie sehr das RIMS den spezifischen Anforderungen des Herstellers genügt,
- wie flexibel der Anbieter auf die Wünsche des Herstellers eingeht,
- welcher Aufwand für die Anbindung an die eigenen Systeme tatsächlich anfällt (z. B. für die Entwicklung von Konnektoren),
- wie kompetent der Support unterstützt und
- wie lange das Projekt dauern wird.
Eine Produkt-Demo ermöglicht es, die Liste der Anbieter einzugrenzen. Sie reicht nicht aus, um die o.g. Fragen zu beantworten.
Auch den Probebetrieb sollte der Hersteller planen. Dazu zählen:
- Fragen entwickeln, die der Probebetrieb beantworten soll (Daraus ergibt sich, welche Produkte, Märkte und Organisationseinheiten beim Probebetrieb berücksichtigt werden.)
- Erfolg- und Abbruchkriterien formulieren (und diese dem Anbieter kommunizieren)
- Finanzielle und personelle Ressourcen planen (die fast immer knapp sind)
- Projektorganisation sicherstellen (Meetings, Steering-Committee etc.)
Tipp 6: Keine Big-Bang-Einführung wählen
Selbst bei einem erfolgreichen Probebetrieb sind Einführungen á la Big Bang keine Empfehlung. Die organisatorischen und technischen Änderungen sind zu umfangreich, um von der Organisation verdaut werden zu können.
Das Gesamtprojekt lässt sich nach verschiedenen Gesichtspunkten zerlegen:
- Zu unterstützende Prozesse
- Betroffene Organisationseinheiten
- Produkte oder Produktklassen
- Märkte (z. B. EU, USA)
- Externe Datenquellen und Organisationen
Tipp 7: Change-Management ernst nehmen
Es klingt banal, kommt aber wegen hohem Zeit- und Kostendruck oft zu kurz:
Sorgfältige Planung und frühe Kommunikation sind entscheidend dafür, dass alle betroffenen Mitarbeiter die Einführung des RIMS verstehen und akzeptieren. Schulungen und Schulungsunterlagen sollten bereitstehen, um die Benutzer mit der neuen Software vertraut zu machen.
Tipp 8: QM-Anforderungen erfüllen
Jedes neue System stößt auf das Interesse der Auditoren. Diese fragen nicht nur nach den Kompetenzen und Schulungsnachweisen, sondern auch nach den überarbeiteten Verfahrensanweisungen (und deren Freigaben) und nach der Validierung des Systems.
Der Artikel Computerized Systems Validation zeigt, wie Software wie ein RIMS zu validieren ist und welche Normen dabei helfen.
Die Aufwände und Zeitpunkte für die Validierung müssen im Projektplan enthalten sein.
Tipp 9: Interne „Evangelisten“ etablieren
Die Wahrscheinlichkeit ist hoch, dass die initiale Begeisterung verfliegt, wenn die ersten Bugs und der ganze Aufwand für das Projekt sichtbar werden.
Daher ist es nützlich, wenn interne Mitarbeitende als kompetente Evangelisten
- das Projekt vorantreiben,
- die Wünsche an die Weiterentwicklung und Anpassung der Software koordinieren,
- die Schnittstelle zum Anbieter bilden und
- als schnell verfügbarer Ansprechpartner bei Fragen und Schwierigkeiten dienen.
Tipp 10: Ausstiegsszenario beschreiben und berechnen
Laut Standish Group erreichen die meisten IT-Projekte nicht innerhalb der vorgegebenen Zeit und Kosten ihre Ziele, oder sie scheitern ganz. Daher gehört es zum Risikomanagement des Herstellers, dieses Scheitern einzuplanen:
- Ausstiegsklauseln in den Verträgen sicherstellen
- Kosten für den Ausstieg berechnen
- Ausstiegsszenarien beschreiben, beispielsweise wie das Unternehmen „zurückmigriert“ bzw. ohne das RIMS oder dessen Anbieter die Prozesse aufrechterhalten werden können
7. Fazit und Zusammenfassung
a) Systeme sind noch keine Lösung
Jeder Hersteller benötigt effektive und effiziente regulatorische Prozesse, um
- seine Produkte schnell und sicher in die Märkte zu bringen,
- die Konformität dieser Produkte und seines Unternehmens fortlaufend zu gewährleisten und
- mit den limitierten Ressourcen (v. a. Regulatory Affairs Experts) auszukommen.
Dafür können Regulatory Information Management Systems (RIMS) eine Lösung sein, sie sind es aber nicht immer. Ein RIMS kann sogar neue Probleme schaffen:
- Zusätzliche Schnittstellen und erhöhte Komplexität der Systeme und Prozesse
- Finanzielle und regulatorische Risiken bei der Einführung des RIMS
- Verankerung in der Vergangenheit durch ein RIMS, das alte Konzepte wie dokumentenbasierte Einreichungen zementiert
Die digitale Transformation Ihres Unternehmens gelingt nicht durch Einführung einer neuen Software. Sie wird sogar behindert durch Software, die auf Konzepten der Vergangenheit basiert, z. B. einem dokumentenorientierten Ansatz.
In diesem Artikel zur digitalen Transformation erfahren Sie, was transformiert werden muss und wie diese Transformation am wahrscheinlichsten gelingt.
b) Voraussetzungen müssen erfüllt sein
Diese digitale Transformation gelingt nur durch
- ein präzises Zielbild künftiger regulatorischer Systeme und technischer Lösungen,
- eine klare Roadmap, um diese Ziele zu erreichen und um mit künftigen regulatorischen Systemen kompatibel zu sein,
- regulatorisches Expertenwissen auf Seiten des Herstellers und des Anbieters, um diese Roadmap umzusetzen (etwa die Prüfung von Technischen Dokumentationen zu automatisieren) und damit den erwarteten Nutzen tatsächlich zu erreichen.
Insbesondere RIMS-Anbieter mit Schwerpunkt im pharmazeutischen Umfeld verfügen oft nicht über das Expertenwissen, das Medizinproduktehersteller benötigen.