
Das Scrutiny-Verfahren stellt eine der großen Änderungen dar, die die neue Medizinprodukteverordnung (MDR) einführt. Dieses Verfahren wird auch als Konsultationsverfahren bezeichnet.
| Inhaltsübersicht |
|---|
| Ziele und Gründe » |
| Ablauf des Scrutiny-Verfahrens » |
| Beteiligte Experten » |
| Konsequenzen des Konsulationsverfahrens » |
| Zusammenfassung, Fazit, Kritik » |
Ziele des Scrutiny Verfahrens und Gründe für dessen Einführung
Sicherheit von Patienten gewährleisten
Der europäischen Kommission lag eines besonders am Herzen, als sie die Medizinprodukterichtlinien (wie die MDD) durch die Medizinprodukteverordnungen ablöste: Sie wollte und will die Sicherheit von Patienten in der Europäischen Union besser gewährleisten als in der Vergangenheit.
Um dieses Ziel zu erreichen, muss sichergestellt werden, dass die Medizinprodukte tatsächlich
- den versprochenen Nutzen erbringen,
- die versprochenen Leistungen erfüllen und
- keine inakzeptablen Risiken bergen.
Das sind genau die Nachweise, die im Rahmen der klinischen Bewertung zu führen sind.
Lesen Sie hier mehr zum Thema Klinische Bewertung und MEDDEV 2.7./1 Revision 4
Auf die „wirklichen“ Experten hören
Um zu beurteilen, ob dieser Nachweis dem Hersteller tatsächlich gelingt, bedarf es Experten. Das Scrutiny-Verfahren sieht vor, genau solche Experten für bestimmte Produkte zu konsultieren. Daher stammt der Begriff „Konsultationsverfahren“.
Die deutsche Übersetzung von „Scrutiny“ lautet „genaue Untersuchung“, „genaue Prüfung“ oder „prüfender Blick“.
Die Medizinprodukterichtlinien sahen die Fachgutachter bei den benannten Stellen noch als die einzigen zwingend einzubeziehenden Experten. Darauf möchte sich die Kommission nicht mehr ausschließlich verlassen. Offensichtlich hat sie kein ausreichendes Vertrauen, dass die benannten Stellen für besonders kritische und innovative Produkte über diese Experten verfügen.
Folglich schreibt die MDR wörtlich: „Bei [bestimmten Produkten] sollten Benannte Stellen […] verpflichtet sein, Expertengremien zu beauftragen, ihre Berichte über die Begutachtung der klinischen Bewertung zu kontrollieren.“
Regulatorische Anforderungen an das Scrutiny Verfahren
Begrifflichkeiten
Die MDR verwendet den Begriff „Scrutiny“ nur ein einziges Mal, nämlich in der Überschrift des Artikels 55 („Mechanism for scrutiny of conformity assessments of certain class III and class IIb devices“). Sonst nutzt sie den Begriff des Konsultationsverfahrens („consultation procedure“).
Dieser Artikel nutzt die beiden Begriffe (Scrunity-Verfahren und Konsultationsverfahren) daher synonym.
Betroffene Produkte
Das Konsultationsverfahren betrifft gemäß Artikel 54 der MDR folgende Produkte:
- implantierbare Produkte der Klasse III und
- aktive Produkte der Klasse IIb, die dazu bestimmt sind, ein Arzneimittel an den Körper abzugeben und/oder aus dem Körper zu entfernen.
Auf das Konsultationsverfahren kann ggf. verzichtet werden. Die Voraussetzung dafür besteht darin, dass die Produkte bereits in ähnlicher Form in den Verkehr gebracht wurden und dass Änderungen das Nutzen-Risiko-Verhältnis nicht beeinträchtigen. Letzteres muss der Hersteller nachweisen.
Ablauf des Scrutiny Verfahrens
Die Hersteller reichen wie üblich ihre Unterlagen der benannten Stelle ein. Die benannte Stelle prüft diese und verfasst „einen Bericht über die Begutachtung der klinischen Bewertung“ („clinical evaluation assessment report“). Diesen Bericht reicht sie der Kommission ein, die ihn wiederum an das/ein Expertengremium weiterleitet (s. Abb. 1).
Das Expertengremium entscheidet dann, ob es ein wissenschaftliches Gutachten („scientific assessment report“) erstellt. Falls es das nicht tut, begründet es seine Entscheidung und informiert über die Kommission die benannte Stelle innerhalb von 21 Tagen. Dann darf die benannte Stelle die Konformitätsbewertung fortsetzen.
Erstellt hingegen das „Expert Panel“ das Gutachten, hat es dazu 60 Tage Zeit. Wenn nach 60 Tagen das Gutachten der benannten Stelle nicht vorliegt, darf letztere die Konformitätsbewertung fortsetzen. Andernfalls muss die benannte Stelle diesen Bericht berücksichtigen. Weicht sie von den Empfehlungen ab, muss sie das begründen. Zudem muss sie der Kommission Unterlagen zur Verfügung stellen wie in Abb. 1 zu sehen.
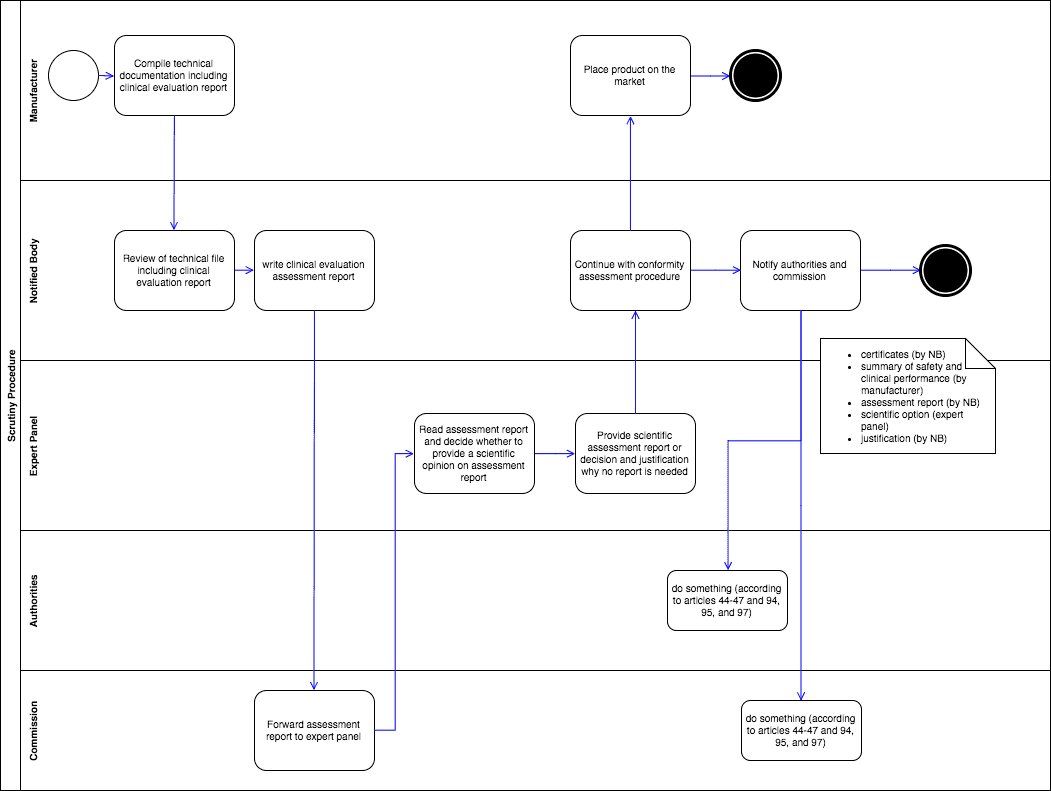
Abb. 1: Das Konsulationsverfahren (Scrutiny-Verfahren) durchläuft benannte Stellen, die EU-Kommission, Expertengremien und (nationale) Behörden. Zum Vergrößern bitte klicken
Anforderungen an die „Experten“
„Die Expertengremien bestehen aus Beratern, die die Kommission auf der Grundlage aktuellen klinischen, wissenschaftlichen oder technischen Fachwissens auf dem betreffenden Gebiet und nach einer geografischen Verteilung berufen hat, die die Vielfalt der wissenschaftlichen und klinischen Konzepte in der Union widerspiegelt“, schreibt Artikel 106 Absatz (3).
„Die Mitglieder der Expertengremien […] dürfen Weisungen von Benannten Stellen oder Herstellern weder einholen noch entgegennehmen. Jedes Mitglied gibt eine Interessenerklärung ab, die öffentlich zugänglich gemacht wird.“
Konsequenzen für die Hersteller
Die erste Konsequenz, die das Scrutiny-Verfahren für die Hersteller bedeutet, liegt auf der Hand: Das Konformitätsbewertungsverfahren dauert länger.
| Arbeitsschritt | Zeitliche Verzögerung |
|---|---|
| Prüfen der Akte bei der benannten Stelle | Theoretisch keine. Allerdings wird eine benannte Stelle sicher noch sorgfältiger arbeiten, wenn sie weiß, dass die Akte danach an ein externes Expertenpanel geht |
| Erstellen des „clinical evaluation assessment reports“ | Dieser Arbeitsschritt ist neu und dürfte im Schnitt eine Woche Verzögerung bedeuten |
| Weiterleiten des Berichts an die Kommission | Vernachlässigbar |
| Weiterleiten des Berichts von der Kommission ans Expertenpanel | Theoretisch vernachlässigbar, da dies „sofort“ erfolgen muss |
| Entscheidung des Expertenpanels über wissenschaftliches Gutachten | Bis zu 21 Tagen (nach Eingang des Auftrags durch die Kommission) |
| Erstellen des wissenschaftlichen Gutachtens durch das Expertenpanel | Bis zu 60 Tagen (nach Eingang des Auftrags durch die Kommission) |
Wir vermuten, dass das Scrutiny-Verfahren die Konformitätsbewertung um ein Vierteljahr verzögern wird.
Dass sich die benannten Stellen den zusätzlichen Aufwand bezahlen lassen, liegt auf der Hand.
Zusammenfassung und Fazit
Die Motivation der EU Kommission ist ehrenwert, bei hochkritischen Produkten auf Experten zu vertrauen. Es ist auch nachvollziehbar, dass die Kommission daran zweifelt, dass jede benannte Stelle zu jedem Produkt über entsprechende Experten verfügt.
Doch es bleiben einige Fragen und auch Zweifel, was die wirklichen Konsequenzen sind, die sich durch das Konsultationsverfahren ergeben:
- Verfügbarkeit von Experten
In gewissen Produktnischen gibt es z.B. im deutschsprachigen Raum nur eine Handvoll Experten. Auf die greifen die benannten Stellen und Hersteller bereits zu. Die Kommission wird keine neuen Experten aus dem Hut zaubern können. Eine Unabhängigkeit und Unparteilichkeit ist bei einem engen Markt kaum zu realisieren.
Und ob sich so hochgefragte und hochbezahlte Experten von einer Behörde (mit den Stundensätzen des öffentlichen Diensts) vor den Karren spannen lassen, steht auf einem anderen Blatt. - Expertise der Experten
Es besteht eher die Gefahr, dass sich in den Expertengremien die Gremienexperten und nicht die Fachexperten finden. Und selbst rein wissenschaftlich arbeitende Experten haben oft einen Horizont des wissenschaftlich Möglichen – aber nicht des technisch Möglichen und des ethisch Sinnvollen. - Auswahl der Produkte
Die MDR sieht für das Scrutiny-Verfahren implantierbare Produkte der Klasse III und aktive Produkte der Klasse IIb vor, die dazu bestimmt sind, ein Arzneimittel an den Körper abzugeben. Sind die anderen Produkte der Klasse III nicht kritisch? Ist wirklich nur die Klasse entscheidend oder vielleicht nicht auch die Bewährtheit von Verfahren? Weshalb keine Nanomaterialien? - Auswirkungen auf die Hersteller
Das Konsultationsverfahren bedeutet wie viele andere Neuerungen, die die MDR eingeführt hat, vor allem eins: Mehr Aufwand, höhere Kosten und einen längeren Zeitpunkt, bis das Produkt vermarktet werden kann. - Auswirkungen auf das Gesundheitssystem
Eine Folgenabwägung von Produkten, die dem Gesundheitssystem nicht oder erst viel später zur Verfügung stehen, lässt sich auch hier vermissen. Der „sichere Zustand“ besteht nicht immer darin, ein Produkt nicht in den Markt zu lassen. Das scheint die Kommission nicht verstanden zu haben. - Umsetzbarkeit
Die Kommunikation und der Datenaustausch zwischen der Kommission, den Expertenpanels und den benannten Stellen soll in wesentlichen Teilen über die EUDAMED realisiert werden. Sie kennen das Fertigstellungsdatum dieser Datenbank noch nicht? Die EU auch nicht. Wie soll dann eine gesetzeskonforme Zusammenarbeit erfolgen?
Das Fazit dieses Artikels lautet nicht, dass das Scrutiny-Verfahren per se ungeeignet sei. Aber es bestehen Zweifel, dass die EU Kommission ihre damit verbundenen Hoffnungen und Ziele in Gänze erreicht; Hoffnungen und Ziele, die auch der BVMED formuliert.
Es bestehen auch Zweifel daran, dass die EU-Kommission eine Nutzen-Risiko-Abwägung dieses Ansatzes ebenso wissenschaftlich und quantitativ vorgenommen hat, wie sie es von den Herstellern bei der klinischen Bewertung verlangt.



Es stellt sich die ewige Frage, wer kontrolliert den Kontrolleur? Ein Überwachungssystem könnte man so lange fortführen, bis auch dem letzten Unternehmen die Mittel fehlen um alle Kontrolleure und Kontrollmaßnahmen zu finanzieren. Es frägt sich zusätzlich ob nicht zu viele Köche den Brei verderben. Es entsteht allmählich der Eindruck, dass die EU-Behörden zu aller erst die Kontrolle über sich selbst verlieren. Wen wundert es da noch, dass sich immer mehr Bürger, Regionen und ganze Länder von dieser Institution abwenden.
mit freundlichen Grüßen
Friedrich Tieber
Hallo Herr Prof. Johner,
ich finde es noch ganz spannend zu wissen, dass der Hersteller (theoretisch) das in Rede stehende Expertengremium bereits in einer frühen Phase mit seiner klinischen Strategie konfrontieren kann um evtl. böse Überraschungen gegen Ende des Konformitätsbewertungsverfahrens zu vermeiden (siehe MDR, Artikel 61 (2)).
Viele Grüße,
Joachim Schneider
Sehr geehrter Herr Schneider,
die Chancen, im Vorfeld mit dem Expertengremium zu kommunizieren, sehe ich als sehr gering. Dafür ursächlich sind:
Ihren Wunsch kann ich gut verstehen. Ich würde primär mit der benannten Stellen versuchen, Einverständnis zu erreichen.
Viele Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner
mittlerweile existiert das Expertenpanel, aber wir haben noch keine Möglichkeit gefunden, Kontakt aufzunehmen.
Haben Sie diesbezüglich mehr Informationen?
Wir würden gern unsere klinische Entwicklungsstrategie mit einem Experten besprechen wollen, da unsere benannte Stelle dies mit der Begründung ablehnt, dass sie nicht beratend tätig werden darf.
Viele Grüße
Heike Hauptmann
Sehr geehrte Frau Hauptmann,
danke für Ihre Frage. Ich antworte gerne so gut ich kann. Weil es viele Expertenpanels geben wird, bin ich nicht ganz sicher, auf welches Panel Sie Bezug nehmen.
Davon abgesehen zählt es nicht zu den Aufgaben von Expertenpanels Hersteller zu beraten. Die Expertenpanels dürfen aber Berater aufnehmen.
Wenn ich den Kontext noch besser verstehen, kann ich möglicherweise einen Experten für Sie finden. Nutzen Sie dafür gerne auch das Micro-Consulting.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Prof. Johner,
ich habe in meiner Anfrage Bezug genommen auf den § 61 (2) der MDR, aus dem hervorgeht, dass der Hersteller seine vorgesehene Entwicklungsstrategie vor der klinischen Bewertung durch ein Expertengremium prüfen lassen kann.
Uns geht es darum, mit einem Experten unsere Entwicklungsprojekte hinsichtlich klin. Bewertung / Prüfung vor Einreichung bei der benannten Stelle auf MDR-Compliance zu prüfen.
Konkret geht es dabei um Implantate der Risikoklasse III für Knie und Hüfte.
Viele Grüße
Heike Hauptmann