
Regelmäßig diskutieren Auditoren und Behörden mit den Medizinprodukteherstellern darüber, was der Stand der Technik, der State of the Art, ist. Diese Diskussionen gewinnen vor dem Hintergrund der neuen EU-Verordnungen an Schärfe.
Den Herstellern drohen Verzögerungen bei den Zulassungen, unnötiges Re-Design der Produkte und aufwändige klinische Prüfungen bzw. klinische Leistungsstudien bei den In-vitro-Diagnostika (IVD).
Die MDR und IVDR bestehen auf dem Stand der Technik. Aber sie definieren nicht, was dieser Stand ist. Schlimmer: Sie verwirren durch eine inkonsistente Verwendung des Begriffs und eine ebenso inkonsistente Übersetzung.
Diese Episode verschafft Klarheit und zeigt im Interview mit Professor Röhrig auf, wie die Zeit minimiert werden kann, um den Stand der Wissenschaft in den Stand der Technik zu überführen.
Diese und weitere Podcast-Episoden finden Sie auch hier.

1. Weshalb es so schwer ist, den Stand der Technik zu bestimmen
Fast alle Hersteller sind bereit, die regulatorischen Anforderungen zu erfüllen und Produkte nach dem „Stand der Technik“ zu entwickeln. Leider legt man ihnen dabei viele Steine in den Weg:
a) Keine harmonisierten Normen
Auf absehbare Zeit wird es für MDR und IVDR keine harmonisierten Normen geben. Diese harmonisierten Normen bildeten in der Vergangenheit meist einen guten Konsens zwischen Herstellern einerseits und Behörden bzw. Benannten Stellen andererseits.
b) Keine Begriffsdefinition
Obwohl die beiden Verordnungen den Begriff „Stand der Technik“ über 20 Mal verwenden und diesen „Stand der Technik“ einfordern, verzichten sie auf die Definition des Begriffs.
Wer hofft, in anderen Quellen Klarheit zu finden, sieht sich mit der zusätzlichen Frage konfrontiert, was der Stand der Wissenschaft ist und wie sich dieser vom Stand der Technik unterscheidet.
c) Inkonsistente Verwendung des Begriffs
Obwohl das Fehlen der Definition bereits schwer genug wiegt, hantieren die EU-Verordnungen (MDR, IVDR) zusätzlich mit Varianten des Begriffs. Es gibt nicht nur
- den „Stand der Technik“, sondern auch
- den „neuesten Stand der Technik“,
- den „allgemein anerkannten Stand der Technik“
- und den „gegenwärtigen Stand der Technik“.
d) Fehlerhafte und inkonsistente Übersetzungen
Selbst wer glaubt, diese Varianten differenzieren zu können, wird sich spätestens überfordert fühlen, wenn er die deutschen und englischen Versionen der Verordnungen vergleicht. So fordert die englische Version beim Plan für die klinische Bewertung: „taking into account the state of the art“. Im Leistungsbewertungplan für IVD soll eine „description of the state of the art“ enthalten sein. In den deutschen Versionen der MDR bzw. IVDR ist man an den entsprechenden Textstellen der Meinung, dass der „neueste Stand der Technik“ zu berücksichtigen sei.
Die deutschen Versionen fordert den „neuesten Erkenntnisstand“ und den „neuesten medizinischen Kenntnisstand“, die englische Version spricht immer vom „state of the art“.
Bei diesen Diskrepanzen handelt es sich nicht um Einzelfälle!
e) Benannte Stellen treffen keine klare Aussage
Auch auf explizite Anfrage konnten oder durften die deutschen Benannten Stellen keine klare Aussage dazu treffen, ob und, falls ja, auf welche Normen bzw. Versionen der Normen die Hersteller bei Zulassung ihrer Produkte referenzieren sollen:
- Unter den Richtlinien harmonisierte Normen
- Normen, die zur Harmonisierung unter der MDR/IVDR vorgesehen sind
- Jeweils neueste Version der Normen
f) EU will keine Definition durch die Industrie
Im Standardisation Request (M/565 vom 15.5.2020) verlangt die EU Kommission explizit im Annex III, Part A, Kapitel 1, dass harmonisierte Normen keine relevanten Begriffe definieren dürfen, wenn diese in der MDR oder IVDR nicht definiert wurden.
Damit öffnet die EU Kommission Tür und Tor zu Diskussionen beim Audit oder der Begutachtung der Technischen Dokumentation, wenn sich der Hersteller auf eine Definition in einer (harmonisierten) Norm bezieht. Der Standardisation Request wurde konsequenterweise von CEN und CENELEC zurückgewiesen.
Weil die Gebrauchstauglichkeit der EU-Webseite so bescheiden ist, können Sie das Dokument auch direkt hier herunterladen:
Danke an Beat Keller für diese wertvolle Ergänzung!
Fazit
Hersteller müssen ihre Produkte nach dem Stand der Technik entwickeln. Diese Produkte müssen diesen Stand erfüllen. Leider lässt man die Hersteller ziemlich alleine bei der Frage, was der Stand der Technik ist und wie man diesen bestimmt.
2. Stand der Technik vs. Stand der Wissenschaft
a) Vorbemerkung
Eine allgemeingültige und branchenunabhängige Definition der beiden Begriffe existiert nicht. Die folgenden Ausführungen beschränken sich daher auf die Domäne der Medizinprodukte und IVD.
b) Definition der ISO 14971:2019
Die ISO 14971 hat in der dritten Ausgabe die fehlende Begriffsdefinition für „Stand der Technik“ ergänzt:
entwickeltes Stadium der technischen Möglichkeiten zu einem bestimmten Zeitpunkt, soweit Produkte, Prozesse und Dienstleistungen betroffen sind, basierend auf entsprechenden gesicherten Erkenntnissen von Wissenschaft, Technik und Erfahrung
ISO 14971:2019
Die Norm ergänzt die Definition um folgenden Hinweis:
Der „Stand der Technik“ umfasst die gegenwärtig und allgemein anerkannte gute Praxis bei Technologie und Medizin. „Stand der Technik“ bedeutet nicht unbedingt die technisch fortgeschrittenste Lösung. Der hier beschriebene „Stand der Technik“ wird mitunter als „allgemein anerkannter Stand der Technik“ bezeichnet.
ISO 14971:2019
Dieser Hinweis ist für Hersteller sehr hilfreich:
- Hersteller können den „Stand der Technik“ mit dem „allgemein anerkannten Stand der Technik“ gleichsetzen und somit unnötige Diskussionen über den Unterschied vermeiden.
- Die Anforderungen bestehen nicht zwingend darin, die „technisch fortgeschrittenste Lösung“ zu erreichen.
c) Produktlebenszyklus und Abgrenzung
Die Entwicklung von Produkten und Technologien durchläuft mehrere Phasen (Quelle):
|
Phase |
Aktivität, Ziel |
Typische Akteure |
Beispiel |
|
Grundlagenforschung |
Neues Wissen generieren, ohne ein spezifisches Ziel im Sinne eines Produkts zu verfolgen |
Universitäten, Großforschung |
Bilderkennung mit Machine Learning (ML) |
|
Angewandte Forschung | Neues Wissen generieren, um die Entwicklung von verbesserten oder neuen Komponenten, Produkten oder Verfahren zu ermöglichen |
Universitäten, Hochschulen | Übertragung dieser Verfahren auf die Onkologie |
|
Experimentelle Entwicklung | Ergebnisse der angewandten Forschung mit bestehenden (bewährten) Technologien kombinieren, um Entwicklungsrisiken zu minimieren, d.h. herauszufinden, wie konkrete Produkte und Verfahren beschaffen sein und entwickelt werden können |
Hersteller, Entwicklungs-Dienstleister | Entwicklung eines MVP |
|
Produktentwicklung | Konkretes Produkt entwickeln oder weiterentwickeln, um dieses zulassen und im Markt erfolgreich sein zu können |
Hersteller, Entwicklungs-Dienstleister |
Entwicklung des konkreten Produkts konform zur MDR bzw. IVDR |
Diese Produkte werden vermarktet, überarbeitet und am Ende des Produktlebenszyklus vom Markt genommen.
Der Stand der Technik entspricht laut ISO 14971 der allgemein anerkannten guten Praxis bei Technologie und Medizin und nicht unbedingt der technisch fortgeschrittensten Lösung. Die technisch fortgeschrittenste Lösung ist die, die die Grenze zwischen Stand der Wissenschaft und Stand der Technik markiert. Ob MDR und IVDR mit „neuestem Stand der Technik“ genau diese Grenze meinen, ist unklar.

c) Beispiel
Das folgende Beispiel illustriert die Unterschiede:
|
Stand |
Verschlüsselung |
|
Stand der Wissenschaft |
Quantenverschlüsselung |
|
Stand der Technik |
Empfehlungen und Schlüssellängen des BSIs (z.B. laut BSI TR-02102-1) |
|
Veralteter Stand |
WEP-Verschlüsselung von WLANs |
d) Probleme
Leider unterscheiden viele Regularien nicht präzise zwischen dem Stand der Technik und dem Stand der Wissenschaft. So scheint beispielsweise die MEDDEV 2.7/1 “state of the art” und “current knowledge” teilweise gleichzusetzen. Letzteres entspricht aber eher dem publizierten Stand der Wissenschaft.
3. Regulatorische Anforderungen an den „State of the Art“
a) Ziele der Regularien
Die Regularien versuchen sicherzustellen, dass die Produkte das höchste Maß an Nutzen und Sicherheit gewährleisten. Neuere Produkte bieten oft einen höheren Nutzen. Da ihre Technologien aber nicht so bewährt sind, ist das Nutzen-Risiko-Verhältnis nicht notwendigerweise besser.
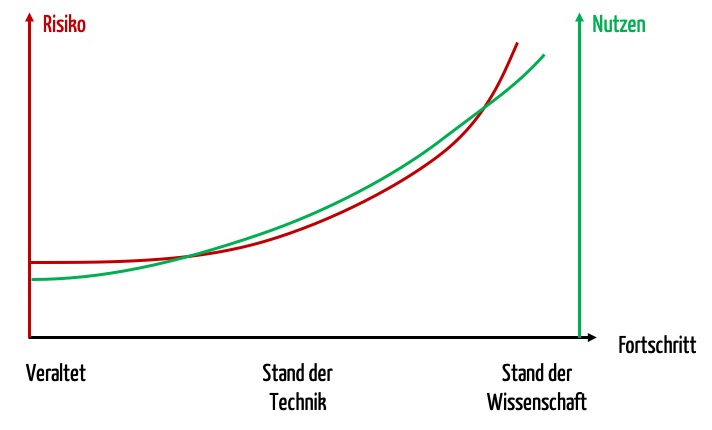
Die Regularien sollten auch gewährleisten, dass die Anforderungen an die Produkte nicht so hoch werden, dass die Hersteller sie nicht mehr wirtschaftlich sinnvoll erfüllen können. Denn Produkte, die zwar ein hervorragendes Nutzen-Risiko-Verhältnis bieten, aber nie auf den Markt kommen, führen zu überhaupt keinem Nutzen.
Ob die Autoren der Regularien diesen zweiten Aspekt wirklich als Ziel verfolgen, erscheint manchmal fraglich.
b) Anforderungen von MDR und IVDR
MDR und IVDR fordern den Stand der Technik u.a. bei Folgendem ein:
- Festlegung der Leistungsanforderungen (MDR Anhang I.1. bzw. IVD Anhang I.9.1)
- Bestimmen des Nutzen-Risiko-Verhältnisses und der Risikoakzeptanz (Anhang I.1.)
- Auswahl risikominimierender Maßnahmen (Anhang I.4)
- Software-Entwicklung (Anhang I.17.2 der MDR bzw. Anhang I.16.2 der IVDR)
- Planung und Durchführung klinischer Bewertungen bzw. Leistungsbewertungen von IVD (Anhang IX.2.1 und Anhang XIII.1)
- Durchführen klinischer Prüfungen (Anhang XV, Kapitel II.3) bzw. klinischer Leistungsstudien für IVD (Anhang XIII.2.3.2)
Die IVDR verlangt den Stand der Technik darüber hinaus bei:
- Planung der Nachbeobachtung der Leistung nach dem Inverkehrbringen (Post-Market Performance Follow-up (PMPF)) (Anhang XIII.5.2)
- Wissenschaftlicher Beratung durch die EU-Referenzlaboratorien (Artikel 100)
c) MEDDEV 2.7/1
Sehr ähnliche Anforderungen formuliert auch die Leitlinie für klinische Bewertungen, die MEDDEV 2.7/1 rev. 4. Auch sie verpflichtet die Hersteller, den State of the Art zu bestimmen und zu berücksichtigen bei:
- Festlegung des Nutzen-Risiko-Verhältnisses
- Aktualisierung der klinischen Bewertung
- Bestimmung der klinischen Leistungsfähigkeit und des „clinical safety profile“
- Literatursuche
Benefits and risks should be specified, e.g. as to their nature, probability, extent, duration and frequency. Core issues are the proper determination of the benefit/risk profile in the intended target groups and medical indications, and demonstration of acceptability of that profile based on current knowledge/ the state of the art in the medical fields concerned.
MEDDEV 2.7/1 rev. 4
When updating the clinical evaluation, the evaluators should verify: compatible with a high level of protection of health and safety and acceptable according to current knowledge/ the state of the art;
Sufficient detail of the clinical background is needed so that the state of the art can be accurately characterised in terms of clinical performance, and clinical safety profile.
Brief summary and justification of the literature search strategy applied for retrieval of information on current knowledge/ the state of the art, including sources used, search questions, search terms, selection criteria applied to the output of the search, quality control measures, results, number and type of literature found to be pertinent. Appraisal criteria used.
Applicable standards and guidance documents.
4. Den Stand der Technik bestimmen
a) Schritt 1: Vorüberlegungen anstellen, erste Kriterien identifizieren
Hersteller müssen den Stand der Technik bestimmen.
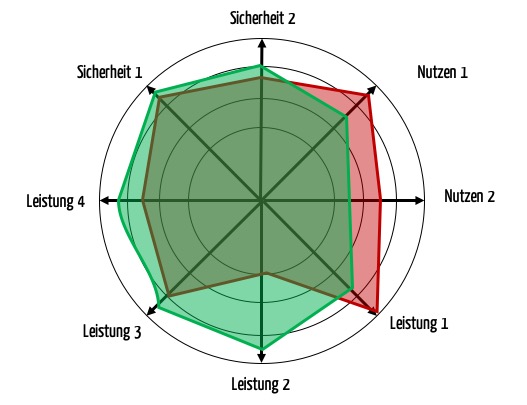
Beachten Sie: Es gibt bei einem Produkt nicht „den“ Stand der Technik! Vielmehr gilt es, den Stand der Technik bezüglich verschiedener Aspekte in den Kategorien klinischer Nutzen, Sicherheit und Leistungsfähigkeit zu bestimmen (siehe Abb. 3).
Die Hersteller sind verpflichtet, die Ergebnisse dieser Analyse in der klinischen Bewertung bzw. Leistungsbewertung für IVD zu dokumentieren. Diese Dokumentation muss auch beschreiben, wie die Hersteller bei dieser Analyse vorgegangen sind.
b) Schritt 2: Leitfragen entwickeln
Hersteller sollten sich Leitfragen überlegen, anhand derer sie den Stand der Technik bestimmen können. Diese Leitfragen lassen sich in zwei Gruppen einteilen:
Alternativen
- Welche alternativen Produkte, z.B. Vorgängerprodukte und Wettbewerbsprodukte, gibt es?
Bei IVD: Sind Referenzmethoden verfügbar und, falls ja, welche sind das? - Existieren alternative Produktkategorien?
(z.B. ein MRT statt eines CTs) - Gibt es alternative Verfahren bzw. gibt es im Fall von IVD alternative Diagnose-Optionen?
(z.B. Chemo- statt Strahlentherapie, qPCR statt Erregerkultur, manuelle Bildauswertung statt ML-gestützte Auswertung) - Welche alternativen Materialien und Technologien stehen zur Verfügung?
- Welche Alternativen bestehen bezüglich des sonstigen Produktdesigns?
- Gibt es andere Produktionsverfahren?
Vergleich der Alternativen
- Wie vergleichen sich diese Alternativen bezüglich des klinischen Nutzens?
Mögliche Aspekte:- Sensitivität und Spezifität einer Diagnose
- Anzahl der Tage im Krankenhaus
- Prozentsatz geheilter Patienten
- Reduktion des Schmerzniveaus auf einer 10-teiligen Skala
- Möglichkeit der Diagnose verschiedener klinischer Subpopulationen
- Wie vergleichen sich die Alternativen bezüglich der Leistungsfähigkeit?
Mögliche Aspekte:- Lebensdauer eines Implantats
- Genauigkeit, mit der ein HF-Impuls erzeugt wird
- Anzahl möglicher Wiederaufbereitungszyklen
- Auflösungsvermögen eines bildgebenden Verfahrens
- Nachweisgrenze einer diagnostischen Analysemethode
- Dauer bis zur verfügbaren Diagnose
- Wie vergleichen sich die Alternativen bezüglich der Sicherheit?
Mögliche Aspekte:- Mean-time-between-failure
- Sicherheit gegen Fehleingaben und andere Nutzungsfehler
- Wahrscheinlichkeit der Detektion von Luftblasen in einem Infusionsschlauch
- Stabilität bei Herunterfallen des Produkts
- Weniger Nebenwirkungen
- Geringere Risiken
c) Schritt 3: Quellen durchsuchen
Leitfäden wie der MEDDEV 2.7/1 schreiben konkrete Informationsquellen vor. Herstellern seien die folgenden Quellen empfohlen:
- Behördendatenbanken, z.B. SwissMedic, BfArM, FDA
- Klinische Fachliteratur, z.B. via PubMed und Embase
- Register
- Messe- und Produktkataloge, Gebrauchsanweisungen
- Technische Datenbanken zu z.B. Materialeigenschaften
- Normen, regulatorische Leitfäden
- Medizinische Leitlinien von Fachgesellschaften
- Informationen der Hersteller über Fehler, z.B. Bug-Reports und Release Notes
- Eigene Post-Market-Daten, z.B. Kundenbeschwerden, Service-Berichte, klinische Nachbeobachtungen bzw. Nachbeobachtung der Leistung für IVD, Log-Dateien
- Labortests
Häufig entsprechen Normen einem Minimalkonsens eines Normengremiums und reflektieren daher nicht immer den Stand der Technik. Beispielsweise dürfte eine Norm wie die IEC 62304 eher als absolute Untergrenze dienen, als dem Anspruch zu genügen, den Stand einer professionellen Software-Entwicklung zu beschreiben.
d) Schritt 4: Quellen auswerten
Zuerst sollten die Hersteller jeden in den Leitfragen adressierten Aspekt einzeln untersuchen. Am Ende dieser Untersuchung sollten die folgenden Fragen beantwortet sein:
- Gibt es Informationen, die Rückschlüsse auf diesen Aspekt erlauben?
- Welche alternativen Produkte, Technologien, Verfahren usw. existieren?
- Welche davon sind überlegen, welche unterlegen?
- Repräsentieren diese Informationen den Stand der Technik oder eher den Stand der Wissenschaft?
Normalerweise dauert die Entwicklung von Normen und Leitfäden mehrere Jahre. Daher repräsentieren diese eher den Stand der Technik, weniger den Stand der Wissenschaft.
Diese Auswertung dient nun als Input, beispielsweise für
- klinische Bewertung bzw. Leistungsbewertung,
- Risikomanagementakte,
- Entwicklung,
- Post-Market Surveillance Plan.
Stellen Sie sicher, dass die alternativen Verfahren, Produkte und Technologien auch wirklich vergleichbar sind. Die Vergleichbarkeit betrifft auch die Patientenpopulation, die Indikationen und Kontraindikationen, die vorgesehenen Anwender und die vorgesehene Nutzungsumgebung.
e) Schritt 5: Analyse periodisch wiederholen
Was heute Stand der Wissenschaft ist, kann in Kürze dem Stand der Technik entsprechen und bald veraltet sein. Beispielsweise dauert dieser Zyklus bei Verfahren des Machine Learnings manchmal nur wenige Monate.
Die Regularien verpflichten die Hersteller daher zur regelmäßigen Aktualisierung. Dies wird u.a. während der klinischen Nachbeobachtung (Post-Market Clinical Follow-up, PMCF) bzw. bei IVD während der Nachbeobachtung der Leistung (PMPF) adressiert. Ein jährlicher Turnus sollte in den meisten Fällen nicht überschritten werden.
5. Fazit und Zusammenfassung
a) Schwierigkeiten und Lösungsansätze
Die EU-Verordnungen machen es den Herstellern durch die fehlende Begriffsdefinition, durch die inkonsistente Verwendung des Begriffs „Stand der Technik“ und durch ärgerliche Übersetzungsfehler unnötig schwer, die zentralen Anforderungen dieser Verordnungen zu erfüllen.
Die ISO 14971 liefert ab der dritten Ausgabe eine dringend benötigte Definition nach. Dass diese Norm nicht für die Harmonisierung vorgesehen ist und damit selbst nicht den Stand der Technik darstellt, wirkt wie ein Treppenwitz.
b) Aspekte, die den Stand der Technik ausmachen
Hersteller sollten folgende Varianten des Begriffs gleichsetzen:
- Stand der Technik
- Allgemein anerkannter Stand der Technik
- Neuester Stand der Technik
Die ISO 14971 erlaubt diese Vereinfachungen.
Die Suche nach alternativen Produkten, Verfahren und Technologien ist dabei unerlässlich. Diese Alternativen müssen die Hersteller bezüglich Sicherheit, Leistungsfähigkeit und klinischem Nutzen vergleichen und bewerten.
Die Aussage, dass ein Produkt dem Stand der Technik entspricht, ist dann gerechtfertigt, wenn sie für alle relevanten Aspekte wie Nutzen, Leistung (z.B. wesentliche Leistungsmerkmale) und Sicherheit zutrifft.
Das bedeutet aber auch, dass ein alternatives Produkt, das dem eigenen Produkt in einem Aspekt überlegen ist, nicht notwendigerweise bedingt, dass ein Hersteller nachziehen muss. Bei dieser Bewertung müssen alle Aspekte betrachtet werden, die den Stand der Technik ausmachen.
c) Rolle der Normen
Bei älteren Normen werden sich Hersteller zunehmend schwertun mit der Argumentation, dass diese den State of the Art abbilden. Bei den jeweils aktuellsten Ausgaben ist diese Vermutung gegeben.
Auch die aktuellsten Normen haben nicht den Anspruch, den Stand der Wissenschaft zu reflektieren.
Leider haben sich die Benannten Stellen noch nicht zu einer eindeutigen Aussage bewegen lassen, ob und wenn ja welche Normen herangezogen werden können oder müssen, um den Stand der Technik zu bestimmen.
d) Ein kontinuierlicher Prozess
Der Stand der Technik ist keine Konstante. Hersteller sind verpflichtet, diesen Stand kontinuierlich zu bestimmen, typischerweise mindestens einmal jährlich. Im Rahmen der Post-Market Surveillance und des Post-Market Clinical Follow-ups bzw. des Post-Market Performance Follow-ups (bei IVD) können Hersteller diese Verpflichtung erfüllen.
Wenn Sie unsicher sind, ob Ihr Produkt dem Stand der Technik entspricht und beim nächsten Audit problemlos durchgehen wird, dann melden Sie sich gerne z.B. über unser kostenloses Micro-Consulting.
Das Johner Institut unterstützt Sie durch das Post-Market Radar dabei, den Stand der Technik automatisiert zu überwachen.
Versionshistorie:
- 2020-09-28: Kapitel 1.f) ergänzt: EU behindert Normen auch durch Verbot relevanter Definitionen



Sehr geehrter Herr Klessascheck,
vielen Dank für den informativen Artikel!
Zu dem Radar-Chart habe ich eine Verständnisfrage: sollen die grüne bzw. rote Fläche den Nutzen bzw. das Risiko eines Produkts (analog den Farben in Abb. 2) oder zwei alternative Produkte im Vergleich darstellen?
Vielen Dank für Ihre Hilfe und beste Grüße
Mareike S.
Liebe Frau Schenk,
dabei handelt es sich um den Vergleich zweier Alternativen, siehe auch Abschnitt b).
Liebe Grüsse, Mario Klessascheck
Hallo,
Vielen Dank für Ihre Ausführungen.
Ein wesentlicher Punkt, bei dem ich doch einige Probleme habe, wird hier nur kurz erwähnt:
Die Regulierung und davon abgeleitete MDCGs verweisen beim Stand der Technik auf Benchmark Devices, sowie performance parameter und deren thresholds:
Wenn ich zwei Produkte von zwei Herstellern habe und es keine allgemein anerkannten Outcome Parameter gibt:
Wie soll ich dies einer vernünftigen Risk Benefit Darstellung zukommen lassen und welche Konsequenzen leiten sich davon ab?
Gesetzt den Fall: Produkt A hat OutcomeXY 90% Produkt B hat Outcome 70% (also schlechter), aber Produkt B hat weniger Nebenwirkungen (5 statt 10% bei Produkt A).
Produkt B wäre hinsichtlich Performance nicht State of the Art, Produkt A nicht state of the art hinischtlich Sicherheit.
Ganz abgesehen davon, dass sich die verfügbare Information hinsichtlich der Outcome und Safety Parameter zwischen Produkt A und B sich unterscheiden (zB für Performance unterschiedliche Tests verwendet wurden).
Reicht dies nur beschreibend widerzugeben, oder muss ich davon Konsequenzen ableiten? Von NBs werden hier ja mittlerweile eindeutige Zahlen und thresholds gefordert, die nur bei direkten head to head studies (wenn überhaupt) erlangbar wären, die Hersteller aus verständlichen Gründen ablehnen (Kriegserklärung ;-), Alle Hersteller von Konkurenzprodukten? Welches (veraltete) Modell, etc) und auch anfällig für Manipulation wären. (ZBsp Hersteller von Produkt A nimmt nur solche Endpunkte in die Studie auf, bei denen er eine Überlegenheit zu Produkt B erwartet).
Wie kann ich in diesem Fall den State of the Art und das davon abgeleitete Risk Benefit (im Hinblick auf benchmark devices) darstellen?
Lieber Herr Schwaiger,
Konzentrieren Sie sich nicht zu sehr auf andere Geräte und unterschieden Sie bei der Analyse zwischen Sicherheit und Leistung. In Bezug auf Sicherheit legen Normen den aktuellen Stand der Technik fest, den Sie direkt übernehmen können. Das verbleibende Risiko ist dann als akzeptabel einzustufen. Die Leistung – insbesondere deren Grenzen – müssen Sie klinisch festlegen und dürfen diese nicht einfach übernehmen (es sei denn es gibt Normen oder Literatur). Die MDR verlangt im Abschnitt klinische Bewertung eine: detaillierte Beschreibung des angestrebten klinischen Nutzens für die Patienten mit relevanten konkreten Parametern für das klinische Ergebnis. Das wichtige Schlagwort ist der Begriff „Parameter„. Klinische Leistung führt zu -> Klinischen Nutzen -> führt zu Clinical outcome parameter. Diese wiederum kommen aus der Klinischen Bewertung. Damit können Sie den Nutzen sehr gut auf messbare Parameter zurückführen.
Sie können den Nutzen auch semi-quantitativ beschreiben, beispielsweise indem Sie die Anzahl der Patienten angeben, die davon profitieren, die Wahrscheinlichkeit des Auftretens des gewünschten Nutzens und eventuell auch die Dauer, über die dieser Nutzen bestehen bleibt. Ebenso ist es möglich, die Restrisiken detailliert zu beschreiben, wie zum Beispiel die Beschreibung der Patientengruppen, die am stärksten von Nebenwirkungen betroffen sind, oder die Anzahl der Patienten, bei denen Nebenwirkungen zu erwarten sind. Auditoren empfehlen, eine Bewertung pro Schadensklasse vorzunehmen, insbesondere im Hinblick auf die höheren Schadensklassen, die meldepflichtig sind. Eine mathematische Berechnung des Verhältnisses von Nutzen zu Risiko ist weder möglich noch gefordert.
Konnte ich Ihnen ein wenig helfen?
Liebe Grüsse, Mario Klessascheck