
Die Branchenanalyse Medizintechnik aus dem Jahre 2020 veranschlagte den Markt für Medizinprodukte in Südkorea auf 6,7 Mrd. USD. Aufgrund einer jährlichen Wachstumsrate der Importe von (geschätzt) 10 % und der zunehmenden Überalterung der Bevölkerung bei gleichzeitigem Anstieg der medizinischen Grundversorgung ist mit einem kontinuierlichen Wachstum des südkoreanischen Markts zu rechnen.
Dieser Beitrag zeigt auf, welche regulatorischen Rahmenbedingungen in Südkorea beim Verkauf von Medizinprodukten eingehalten werden müssen und welche Hürden es zu bewältigen gibt.
Regulatorische Rahmenbedingungen
a) Verantwortlichkeit
Seit 2017 ist für die Regulierung und Zulassung von Medizinprodukten in Südkorea das MFDS (Ministry of Food and Drug) zuständig. Das MFDS ist für die Registrierung von Medizinprodukten, die Definition von Regularien und das Ausstellen von Zertifikaten zuständig.
b) Regularien
Den Rechtsrahmen für die Registrierung und den Vertrieb von ausländischen Medizinprodukten in Südkorea bildet der Medical Device Act für Medizinprodukte und der Act On In vitro Diagnostic Medical Devices für In-vitro-Diagnostika.
Die Gesetzestexte dienen folgendem Ziel und Zweck (entnommen aus der englischen Übersetzung):
- The purpose of this Act [Medical Device Act] is to promote the efficient management of medical devices and further contribute to the improvement of public health by providing for matters concerning the manufacturing, import, distribution, etc. of medical devices. (Medical Device Act).
- The purpose of this Act [Diagnostic Medical Devices] is to improve public health and contribute to the advancement of in vitro diagnostic medical devices through ensuring safety, improving quality, and strengthening international competitiveness, of such devices by providing for matters necessary for handling, such as manufacturing and import, and management of, and support for the devices (Act On In Vitro Diagnostic Medical Devices).
c) Qualifizierung als Medizinprodukt in Südkorea
Die Definition des Begriffes Medizinprodukt ist dem Artikel 2 des Medical Device Act zu entnehmen. Dieser definiert ein Medizinprodukt wie folgt:
Der Begriff Medizinprodukt bezeichnet ein Instrument, eine Maschine, ein Gerät / einen Apparat, ein Material, eine Software oder ein anderes ähnliches Produkt, das allein oder in Kombination für Menschen und Tiere zu folgenden Zwecken verwendet wird:
– Zum Zwecke der Diagnose, Heilung, Linderung, Behandlung und Vorbeugung von Krankheiten
Medical Device Act, Art. 2
– Zum Zwecke der Diagnose, Heilung, Linderung oder Korrektur einer Verletzung oder Beeinträchtigung
– Zum Zwecke des Testens, Ersetzens bzw. Umwandelns einer Struktur oder Funktion
– Zum Zwecke der Empfängnisverhütung
Es gibt mehrere Unterschiede zur Verordnung (EU) 2017/745 über Medizinprodukte (MDR):
- Die Ausweitung des Begriffes auf die Verwendung bei Tieren (die MDR schließt nur Menschen ein)
- Die pharmakologische oder immunologische Hauptwirkung des Produkts (beschrieben in der MDR) ist nicht Bestandteil der koreanischen Begriffsdefinition.
- Die Gewinnung von Informationen durch In-vitro-Untersuchungen ist nicht Bestandteil der koreanischen Begriffsdefinition „Medizinprodukt“.
Begriffsdefinitionen hinsichtlich In-vitro-Diagnostika sind dem Act on In vitro Diagnostic Medical Devices zu entnehmen.
Die Schritte zur Produktregistrierung in Südkorea
Der Registrierungsprozess für ausländische Medizinprodukte in Südkorea stellt die Abbildung 1 dar. Als Ziel dieses Prozesses steht die Zulassung bzw. Ausstellung einer Pre-Market-Zulassungslizenz für Ihre Produkte durch die südkoreanischen Behörden und die damit verbundene Möglichkeit des Imports und Vertriebs Ihrer Produkte auf dem südkoreanischen Markt.

Erhaltene Pre-Market-Zulassungslizenzen müssen alle fünf Jahre erneuert werden.
Schritt 1: Benennung des Korean Licence Holder KLH
Vor Beginn des Registrierungsprozesses ihrer Medizinprodukte müssen Hersteller ohne Niederlassung in Südkorea einen koreanischen Lizenzinhaber (KLH – Korean Licence Holder) im Land benennen und einen Vertrag mit diesem abschließen. Der KLH übernimmt die Funktion des rechtlichen Vertreters gegenüber dem Ministerium (MFDS).
Folgende Voraussetzung müssen Korean Licence Holder erfüllen:
- In Südkorea ansässig sein und einen eingetragenen Sitz in Südkorea haben
- Als KLH beim MFDS mit Lizenz für Medizinprodukte und In-Vitro-Diagnostika registriert sein
- Als Qualitätsmanager beim MFDS registriert sein
Der Korean Licence Holder wird unabhängig von der Risikoklasse der Medizinprodukte oder In-Vitro-Diagnostika benötigt.
Gemeinsam mit unserem Partner vor Ort unterstützen wir Sie als KLH und bei allen Fragen rund um die dortige Medizinproduktregulierungen. Nehmen Sie einfach Kontakt mit uns auf.
Schritt 2: Klassifizierung des Medizinprodukts
Das MFDS verwendet ein vierstufiges Klassifizierungssystem für Medizinprodukte, das auf dem Risiko für den menschlichen Körper basiert. Es folgt hiermit dem Ansatz der Richtlinie Principles of Medical Devices Classification der GHFT (Global Harmonization Task Force) bzw. den analogen IMDRF-Regeln.
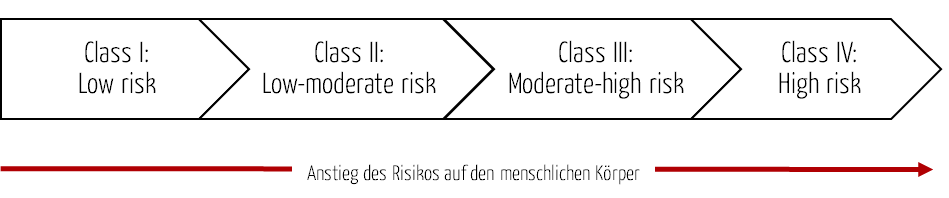
| Klasse | Risiko | Beispiel |
| Klasse I | Medizinprodukte mit geringem Risiko | Pinzette, Stethoskope, Patientenbett (manuelle Betätigung) |
| Klasse II | Medizinprodukte mit geringem bis mittlerem Risiko | Spritze, Infusionspumpe, Patientenbett (elektrische Betätigung) |
| Klasse III | Medizinprodukte mit mittlerem bis hohen Risiko | Anästhesie-System, Beatmungsgerät, Dialyse-System |
| Klasse IV | Medizinprodukte mit hohem Risiko | Herzklappe, Koronarstent, Bypass-Systeme |
Als erstes Hilfsmittel für die Klassifizierung von Produkten kann die Datenbank Medical Device Products and their Assigned Classes des MFDS herangezogen werden. In regelmäßigen Abständen veröffentlicht das MFDS in der Datenbank eine aktualisierte Übersicht von Klassifizierungen registrierter Medizinprodukte, inklusive Beispiele, Beschreibungen und Kategorienummern.
Nutzen Sie als weitere Hilfsmittel zur Einteilung Ihrer Produkte in eine der vier Risikoklassen die Klassifizierungsregeln der GHTF-Richtlinie Principles of Medical Devices Classification (Kapitel 8.0).
Schritt 3: Zertifizierung nach Korean Good Manufacturing Practices
Ausländische Hersteller von Medizinprodukten (auch In-vitro-Diagnostika) müssen ihr QM-System vor Start der jeweiligen Produktregistrierung hinsichtlich KGMP (Korean Good Manufacturing Practices) zertifizieren lassen.
Mittels KGMP-Zertifizierung soll sichergestellt werden, dass die Medizinprodukte gemäß den Anforderungen und Qualitätsstandards der MFDS produziert, kontrolliert und nachbeobachtet werden. Detaillierte Informationen zur Implementierung der KGMP gibt die Regulation on Good Manufacturing Practices (GMP) for Medicinal Products.
Von der Zertifizierung nach KGMP sind die Produkte der Klassen II bis IV betroffen. Medizinprodukte der Klasse I sind von dieser Pflicht ausgenommen.

Den Antrag auf KGMP-Zertifizierung stellt der Korean Licence Holder (KLH), den der ausländische Hersteller des Medizinprodukts gewählt und vertraglich gebunden hat, beim MFDS.
Die KGMP-Zertifizierung ist mit dem gewählten KLH (Korean Licence Holder) verknüpft. Bei Wechsel des KLH ist eine Erneuerung des KGMP-Zertifikats notwendig.
Bei der Neuregistrierung wird die Prüfung als Vor-Ort-Audit der Produktionsstätte durchgeführt. Die Kosten liegen je nach Umfang zwischen 4.000 und 6.000 USD. Es ist hierbei mit einer Vorlaufzeit von mehreren Monaten zu rechnen.
Bei Produktregistrierung mit gültigem KGMP-Zertifikat oder bei Verlängerung des Zertifikats wird eine Dokumentenprüfung durchgeführt. Die Kosten liegen zwischen 1.000 und 1.500 USD. Es ist mit einer Dauer von einigen Wochen zu rechnen.
Das KGMP-Zertifikat behält drei Jahre seine Gültigkeit und muss anschließend verlängert werden.
Trotz Anlehnung an die ISO 13485 ersetzt eine Zertifizierung nach ISO 13585 nicht das KGMP-Zertifikat. Es müssen zusätzliche Anforderungen eingehalten bzw. umgesetzt werden, z. B. an die Post-Market Surveillance und das Meldewesen.
Schritt 4: Produktregistrierung
Die „Zulassungsverfahren“ von Medizinprodukten in Südkorea (auch als Registrierung oder Zertifizierung von Medizinprodukten definiert) unterscheiden sich je nach festgelegter Medizinproduktklasse (I bis IV). Die unterschiedlichen Verfahren werden im Folgenden näher beschrieben.
Alle für die Registrierung bzw. Zertifizierung oder Zulassung Ihrer Produkte zu erstellenden Dokumente und notwendigen Informationen müssen in Koreanisch bereitgestellt werden!
Registrierung von Produkten der Klasse I (4a)

Hersteller von Medizinprodukten der Klasse I stellen für die Registrierung ihrer Produkte einen Antrag auf Product Notification beim NIDS (National Institute of Medical Device Safety Information).
Nach erfolgreichem Upload des kompletten Antrags einschließlich aller Informationen kann der Hersteller die Produktregistrierung seiner Medizinprodukte der Klasse I eigenständig abschließen. Dieser Vorgang ist mit Kosten von ca. 75 USD verbunden.
Medizinprodukte der Klasse I sind von einer KGMP-Zertifizierung befreit.
Zertifizierung bei Produkten der Klasse II (4b)
Die Zulassungs- bzw. Zertifizierungsverfahren der Produkte der Klasse II unterscheiden sich hinsichtlich der Substantial Equivalency (SE). Die Produkte werden nach den Regularien des MFDS in vier Kategorien unterteilt:
- Recognized SE (Substantial Equivalency) Devices
- SE (Substantial Equivalency) Devices
- Modified Device
- New Devices
Südkorea unterscheidet nicht nur die Klassen I bis IV, sondern innerhalb der Klasse II nochmals die Kategorien 1 bis 4.
Produkte der Kategorien 1 bis 3 benötigen eine Produktzertifizierung durch das NIDS. Hierbei ist (analog zu den Produkten der Klasse I) beim NIDS ein Antrag auf Product Notification zu stellen. Dieser ist durch zusätzliche Dokumente zu ergänzen.

Bei Produkten der Kategorie 1 (Recognized SE Devices) ist zusätzlich zum Antrag auf Product Notification ein Bericht über die Sicherheit und Leistung (Test Report of Safety and Performance) zu erstellen. Es ist mit einer Bearbeitungszeit von fünf Tagen und Kosten von ca. 120 USD für die Zertifizierung zu rechnen.
Produkte der Kategorie 2 (SE Devices) und der Kategorie 3 (Modified Devices) durchlaufen ergänzend zum Antrag auf Product Notification eine detaillierte Prüfung der einzureichenden Dokumente. Für die Prüfung der zusätzlichen Dokumente ist ergänzend mit einer Bearbeitungszeit von 25 Tagen und Kosten von ebenfalls ca. 120 US-Dollar zu rechnen.
Bei Produkten der Kategorie 2 (SE Devices) ist ergänzend zu den Informationen der Produkte der Kategorie 1 eine Bewertung von Vergleichsdaten (Comparison Data) zu bereits auf dem Markt befindlichen Produkten zu erstellen.
Bei Produkten der Kategorie 3 (Modified Devices) ist ergänzend zu den Informationen der Produkte der Kategorie 2 eine Dokumentation des Entstehungs- und Entwicklungsprozesses (Origin & Development Process) inkl. Nutzungsstatus im Ausland (Usage Status Overseas) zu erstellen.
Zulassung von Produkten der Klasse II (New Devices) und der Klassen III bis IV (4c)
Bei Medizinprodukten der Klasse II in der Kategorie 4 (New Devices) sowie bei den Produkten der Klassen III und IV wird ein Zulassungsantrag beim MFDS gestellt.
Für Hersteller gibt es hierbei zwei Optionen:
- Die generelle Prüfung der Technischen Dokumentation
- Die Prüfung der Sicherheit und Wirksamkeit des Produkts (SER-Akte)
Für die Prüfung der Dokumente ist mit einer Bearbeitungszeit von 60 bis 80 Tagen und Kosten von zwischen 600 und 1.200 USD (abhängig von der Medizinproduktklasse) zu rechnen.

Zusätzlich zum Zulassungsantrag und der Technischen Dokumentation bzw. dem SER sind weitere relevante Informationen anzugeben bzw. einzureichen:
- Alle relevanten Testberichte (Validierungsdokumentation)
- Vergleichsdaten zu auf dem Markt befindlichen Produkten (Comparison Data)
- Beschreibung der Wirkweise bzw. des Funktionsprinzips des Produkts (Mode of Action)
- Prozessbeschreibungen der Sicherheit und Leistung des Produkts (Safety & Performance Process)
- Berichte zu klinischen Studien (ausgenommen Produkte, die eine SER-Prüfung durchlaufen)
- Informationen zum Nutzungsstatus im Ausland
Bei Medizinprodukten der Klasse IV müssen Hersteller die Technische Dokumentation im STED-Format einreichen. Bei den Klassen II (New Devices) und III ist die Einreichung im STED-Format freiwillig.
Ist in der Technischen Dokumentation keine ausreichende Äquivalenz zu einem auf dem Markt befindlichen Produkt (Comparison Data) zu ermitteln, muss das Produkt ergänzend zur Prüfung der Dokumentation einen Clinical Data Review (CDR) durchlaufen.
Ausländische Prüfberichte werden in Südkorea nur in Teilen für die Produktzulassung akzeptiert. Es empfiehlt sich daher, vorab die relevanten Berichte hinsichtlich Anerkennung prüfen zu lassen.
Fazit und Zusammenfassung
Der südkoreanische Markt stellt aufgrund der jährlich wachsenden Importrate von Medizinprodukten einen interessanten Markt für ausländische Hersteller dar. Hierbei gibt es jedoch einige Hürden zu überwinden:
- Ausländische Hersteller benötigen unabhängig der Medizinproduktklasse einen vor Ort ansässigen Korean Licence Holder (KLH).
- Es müssen die Anforderungen der KGMP (Korean Good Manufacturing Practices) eingehalten und zertifiziert werden.
- Alle notwendigen Dokumente und Akten sind in Koreanisch einzureichen.
- Ausländische Prüfberichte werden nur in Teilen anerkannt.
Die Höhe der Hürden hängt von der Klasse des Produkts sowie bei Produkten der Klasse II von der Kategorie des Produkts ab.
Gemeinsam mit unserem Partner vor Ort können wir Ihnen helfen, diese Hürden überwinden. Wir unterstützen Sie gerne bei Ihrem Registrierungsprozess. Schreiben Sie uns einfach an.


Guten Tag,
in welcher Verordung wird nun der Aufbau der TD konkret bescheiben (vergleichbar mit ANNEX 2 der MDR)
Viele Grüße
Max Schwanau
Sehr geehrter Herr Schwanau,
vielen Dank für ihre spannende Anfrage. Anforderungen an den Aufbau der Technischen Dokumentation sind nur für Produkte der Klasse IV geregelt. Die Technische Dokumentation muss im STED-Format eingereicht werden (https://www.johner-institut.de/blog/regulatory-affairs/sted-technische-dokumentation/). Für Produkte der Klassen III und II ist diese freiwillig. Zudem sind der Technischen Dokumentation aller drei Klassen relevante Prüfberichte beizulegen.
Mit freundlichen Grüßen,
Florian Krafft