
Die europäische Gesetzgebung definiert Systeme und Behandlungseinheiten („systems and procedure packs“) und unterscheidet verschiedene Konstellationen. Die regulatorischen Anforderungen an die Hersteller hängen stark von diesen Konstellationen ab.
Lesen Sie in diesem Artikel, was die Gesetzgeber unter Systemen und Behandlungseinheiten verstehen, was die wichtigsten gesetzlichen Anforderungen an die Hersteller sind und welche typischen Fehler Sie vermeiden sollten.
1. Systeme und Behandlungseinheiten: Was versteht man darunter?
a) Definitionen
Sowohl das Medizinproduktegesetz (MPG) als auch die Medizinprodukterichtlinie (MDD) stellen Anforderungen an die Inverkehrbringung von Systemen und Behandlungseinheiten. Aber beide definieren die Begriffe nicht. Dafür holt die EU-Medizinprodukteverordnung (MDR) – nicht zu verwechseln mit der deutschen Medizinprodukteverordnung (MPV) – dieses Versäumnis nach:
„System“ bezeichnet eine Kombination von Produkten, die zusammen verpackt sind oder auch nicht und die dazu bestimmt sind, verbunden oder kombiniert zu werden, um einen spezifischen medizinischen Zweck zu erfüllen;
Quelle: MDR, Artikel 2 (11)
Die MDR definiert auch den Begriff „Behandlungseinheit“:
„Behandlungseinheit“ bezeichnet eine Kombination von zusammen verpackten und in Verkehr gebrachten Produkten, die zur Verwendung für einen spezifischen medizinischen Zweck bestimmt sind;
Quelle: MDR Artikel 2 (10)
Die Definitionen machen klar, dass es einen Überlapp von Systemen und Behandlungseinheiten gibt (s. Abb. 1).
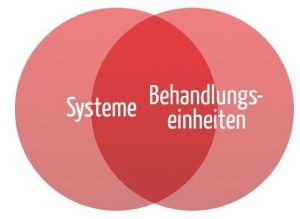
b) Beispiele für Systeme und Behandlungseinheiten
Beispiele von Kombinationen von Produkten, die Systeme und Behandlungseinheit sind:
- HF-Chirurgiegerät, das mit HF-Elektroden zusammen verpackt in Verkehr gebracht wird
- Implantat, das mit dem Knochenzement zusammen verpackt in Verkehr gebracht wird
- Spritzenpumpe, die mit einigen Disposable-Spritzen zusammen verpackt in Verkehr gebraucht werden
Zu den Beispielen für Kombinationen von Produkten, die ein System, aber keine Behandlungseinheit sind, zählen:
- Alle o.g. Beispiele, wenn die Produkte einzeln verkauft, aber (gemäß der Zweckbestimmung) verbunden oder kombiniert werden (sollen)
Beispiele von Kombinationen von Produkten, die eine Behandlungseinheit, aber kein System sind:
- Blutdruckmessgerät, das zusammen verpackt mit dafür geeignetem Desinfektionsmittel in Verkehr gebracht wird
- Infusionspumpe (Medizinprodukt) und Software zum Parametrieren von Infusionspumpen (Zubehör), die gemeinsam verpackt in Verkehr gebracht werden
c) KIS als System oder Behandlungseinheit?
Ein EUGH-Urteil besagt, dass Krankenhaus-Informationssysteme (KIS) Nicht-Medizinprodukte sein können, die jedoch Module enthalten dürfen, die wiederum als Medizinprodukt klassifiziert sind. In diesem Kontext stellt sich die Frage, ob solch ein KIS einem System oder einer Behandlungseinheit im Sinne der gesetzlichen Definition entspricht.
Ein ausführliches Gutachten analysiert, ob Krankenhaus-Informationssysteme als Systeme und Behandlungseinheiten zu betrachten sind.
d) Abgrenzungen zu Kombinationsprodukten
Kombinationen von Medizinprodukten und Arzneimitteln oder biologischen Produkten zählen als „Kombinationsprodukte“ und fallen nicht unter die Definition von „Systeme und Behandlungseinheiten“.
Der Artikel über Kombinationsprodukte beschreibt die Kombination aus Medizinprodukten und Arzneimitteln sowie die regulatorischen Anforderungen daran.
e) Abgrenzung zu Systemen gemäß IEC 60601-1
Auch die IEC 60601-1 spricht von Systemen, genau von „medizinischen elektrischen Systemen“, kurz von ME-Systemen. Sie definiert diese wie folgt:
„Kombination von einzelnen Geräten, wie vom Hersteller festgelegt, von denen mindestens eines ein ME-Gerät sein muss und die durch eine Funktionsverbindung oder durch den Gebrauch einer Mehrfachsteckdose zusammengeschlossen sind“
Quelle: IEC 60601-1 Kapitel 3.64
ME-Systeme werden im Gegensatz zu Systemen gemäß MDR nicht notwendigerweise vom Hersteller als „System“ ausgeliefert. Vielmehr setzen oft die Betreiber die ME-Systeme zusammen.
Liefert der Hersteller ein ME-System aus, kann dies in Form eines Medizinprodukts geschehen, das aus mehreren „Komponenten“ besteht, oder als System im Sinne der Medizinprodukteverordnung MDR.
f) Was keine Systeme und Behandlungseinheiten sind
Die MDCG hat die Leitlinie MDCG 2018-03 (hier der Link zur MDCG 2018-03 rev. 1) veröffentlicht. Darin gibt die MDCG einerseits Hilfestellung zur Vergabe von UDIs für Systeme und Behandlungseinheiten. Sie bestimmt darin aber auch, was sie nicht als System und Behandlungseinheit versteht:
Based on a request of a client or hospital, a natural or legal person in the supply chain may make available together different products, including CE marked devices, which are – in that entire combination – neither placed on the market by that natural or legal person, nor intended by that natural or legal person to be used together for a specific medical purpose. Devices made available in the described manner are not considered as systems or procedure packs.
MDCG 2018-03
Das heißt: Die Entscheidung darüber, ob das gemeinsame Inverkehrbringen von Produkten dazu führt, das diese als System oder Behandlungseinheit zu verstehen sind, hängt auch davon ab, ob der „Inverkehrbringer“ auch Inverkehrbringer eines der einzelnen Produkte dieses Systems oder dieser Behandlungseinheit ist.
Offensichtlich möchte die EU eine Vereinfachung für Firmen erreichen, die „nur“ Produkte in den Verkehr bringen, z. B. als „Convenience Kits“. Als Beispiel nennt die MDCG:
Example: a distributor supplies, upon request of a client, in one shipment, sterile tweezers, a sterile needle and surgery gloves.
MDCG 2018-03
2. Welche regulatorischen Anforderungen sind zu beachten?
Die europäische Rechtsprechung kennt zum einen die Medizinprodukterichtlinien, die in nationales (Medizinprodukte-)Gesetz überführt werden müssen, und zum anderen die EU-Medizinprodukteverordnungen (MDR, IVDR). Dieses Kapitel stellt die regulatorischen Anforderungen vor, die Hersteller erfüllen müssen, die Systeme oder Behandlungseinheiten in den Verkehr bringen.
- Medizinprodukterichtlinie MDD
- Medizinproduktegesetz MPG
- Deutsche Medizinprodukteverordnung MPV
- EK-Med
- EU-Medizinprodukteverordnung MPG
a) Medizinprodukterichtlinie (veraltet)
Die Medizinprodukterichtline MDD forderte in Artikel 12, dass die Hersteller von Systemen und Behandlungseinheiten eine Erklärung abgeben, die besagt, dass sie (die Hersteller)
die gegenseitige Vereinbarkeit der Produkte entsprechend den Hinweisen der Hersteller geprüft und die Arbeitsschritte entsprechend den Hinweisen durchgeführt haben;
das System oder die Behandlungseinheit verpackt und sachdienliche Benutzerhinweise, einschließlich der einschlägigen Hinweise der Hersteller, gegeben haben;
die gesamte Tätigkeit in geeigneter Weise intern überwacht und kontrolliert wurde.
Die MDD sprach nicht vom Hersteller. Dass sich die Anforderungen an Hersteller wenden, ergibt sich erst mittelbar aus der deutschen Umsetzung in MPG § 10(1) und der MPV § 7(6). Erst letztere spricht vom Hersteller, der die Erklärung auszustellen habe.
Die Hersteller müssen für diese Systeme und Behandlungseinheiten keine (erneute) Konformitätsprüfung durchlaufen. Der Gesetzgeber setzt aber voraus, dass „die Produkte eine CE-Kennzeichnung tragen [und] entsprechend ihrer Zweckbestimmung […] zusammengesetzt“ und verwendet werden.
Die MDD definierte „Produkte“ wie folgt:
„Medizinprodukte und Zubehör werden nachstehend „Produkte“ genannt.“
MDD Artikel 1
Damit sprach die MDD nur von Medizinprodukten und Zubehör, nicht von In-vitro-Diagnostika.
Sobald die Kombination ein Produkt enthält, das nicht gemäß der Zweckbestimmung verwendet wird, oder gar eines, das nicht konform mit der MDD in Verkehr gebracht wurde, darf diese Kombination (ohne Konformitätsbewertungsverfahren) nicht als System oder Behandlungseinheit in Verkehr gebracht werden.
Die Hersteller mussten die Erklärungen „für die zuständigen Behörden über einen Zeitraum von fünf Jahren zur Verfügung zu halten“. Genaueres regelt das Medizinproduktegesetz mit seinen nationalen Verordnungen.
Offensichtlich waren sich die Behörden selbst nicht sicher, wann eine solche Erklärung abgegeben werden muss. Ein Regierungspräsidium schrieb an einen Kunden des Johner Instituts:
Wir haben den Vorgang noch einmal geprüft und auch in Rücksprache mit den anderen Regierungspräsidien beschlossen, dass wir eine zusätzliche Meldung als Systemzusammensteller zur Herstelleranzeige nicht akzeptieren.
Grund ist, dass die Kompatibilität der Zubehörsoftware zur Hauptsoftware im Rahmen Ihres Konformitätsbewertungsverfahrens ja bereits nachgewiesen sein muss. Eine Anzeige als Verantwortlicher für das Zusammensetzen von Systemen oder Behandlungseinheiten halten wir nur dann für erforderlich, wenn Sie für mindestens ein Medizinprodukt des Systems nicht Hersteller im Sinne des MPG sind.
Auf welcher Rechtsgrundlage diese Einschätzung erfolgte, erschließt sich aus dem Text nicht. Die großzügige Auslegung erscheint dennoch schlüssig und dürfte im Sinn vieler Hersteller sein.
b) Medizinproduktegesetz
Das deutsche Medizinproduktegesetz überführte den Artikel 12 der MDD durch den § 10 MPG in deutsches Recht. Der entsprechende Gesetzestext lautet:
(1) Medizinprodukte, die eine CE-Kennzeichnung tragen und die entsprechend ihrer Zweckbestimmung innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen zusammengesetzt werden, um in Form eines Systems oder einer Behandlungseinheit erstmalig in den Verkehr gebracht zu werden, müssen keinem Konformitätsbewertungsverfahren unterzogen werden. Wer für die Zusammensetzung des Systems oder der Behandlungseinheit verantwortlich ist, muss in diesem Fall eine Erklärung nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1 abgeben.
Das MPG untersagte im Absatz 4, dass die Systeme und Behandlungseinheiten mit einer zusätzlichen CE-Kennzeichnung versehen werden, und verlangte, dass notwendige Informationen vom Inverkehrbringer der Systeme bzw. Behandlungseinheiten mitgeliefert werden.
Auch das MPG betonte, dass nur dann auf ein Konformitätsbewertungsverfahren für ein System oder eine Behandlungseinheit verzichtet werden darf, wenn dieses ausschließlich Medizinprodukte umfasst, die im Rahmen der ursprünglichen Zweckbestimmung eingesetzt werden.
Das MPG konkretisierte die Anzeigepflicht den Behörden gegenüber im § 25 „Allgemeine Anzeigepflicht“:
(2) Wer Systeme oder Behandlungseinheiten nach § 10 Abs. 1 zusammensetzt oder diese sowie Medizinprodukte nach § 10 Abs. 3 sterilisiert und seinen Sitz in Deutschland hat, hat der zuständigen Behörde unter Angabe seiner Anschrift vor Aufnahme der Tätigkeit die Bezeichnung sowie bei Systemen oder Behandlungseinheiten die Beschreibung der betreffenden Medizinprodukte anzuzeigen.
Demnach bestand für Verantwortliche für das Zusammensetzen von Systemen oder Behandlungseinheiten, sog. Assembler, eine Anzeigepflicht bei der zuständigen Behörde.
Die Hersteller mussten die Anzeige beim Deutschen Institut für Medizinische Dokumentation und Information (DIMDI) tätigen. Sie wurde nach der Erfassung an die zuständige Landesbehörde zur Registrierung weitergeleitet.
Wir hatten uns vom Gewerbeaufsichtsamt der Regierung der Oberpfalz bestätigen lassen, dass die Hersteller Systeme und Behandlungseinheiten beim DIMDI über dessen Webseite anzeigen müssen.
c) Nationale Medizinprodukteverordnung (MPV)
Der o.g. § 10 MPG verwies auf die entsprechende Rechtsverordnung nach § 37 des Gesetzes. Diese Verordnung war die Medizinprodukteverordnung (MPV) in der Fassung vom 27.09.2016 inklusive des § 7 „Konformitätsbewertungsverfahren für die sonstigen Medizinprodukte“:
(6) Für Systeme und Behandlungseinheiten nach § 10 Abs. 1 des Medizinproduktegesetzes hat der Hersteller die Erklärung nach Artikel 12 Abs. 2 Satz 1 der Richtlinie 93/42/EWG auszustellen. Die Erklärung ist mindestens fünf Jahre und im Falle von implantierbaren Produkten mindestens 15 Jahre aufzubewahren. Für Systeme und Behandlungseinheiten nach § 10 Abs. 2 des Medizinproduktegesetzes gelten die Vorschriften der Absätze 1 bis 4 entsprechend.
d) EK-MED
Auch der EK-MED diskutierte das Thema Systeme und Behandlungseinheiten, u. a. im Dokument 3.10 A 15. Dabei geht er darauf ein, was passiert, wenn die Produkte zwar alle ein CE-Zeichen tragen und im Sinne der Zweckbestimmung eingesetzt werden, aber Verfahren (z. B. beim Verpacken) angewendet werden, die die Produktqualität beeinflussen.
e) EU-Medizinprodukteverordnung (MDR)
Die europäische Medizinprodukteverordnung MDR führt die Konzepte der MDD weiter. Sie enthält dazu Artikel 22 „Systeme und Behandlungseinheiten“ und Artikel 29 „Registrierung von Produkten“. Die MDR regelt auch den Umgang mit der UDI.
Artikel 22 „Systeme und Behandlungseinheiten“ Absatz (1)
Der Artikel 22 der MDR entspricht dem Artikel 12 der MDD. Allerdings öffnet er in Absatz (1) c) die Türe für „sonstige Produkte“ und für In-vitro-Diagnostika.
Natürliche oder juristische Personen, die Produkte mit einer CE-Kennzeichnung mit folgenden anderen Medizin- oder sonstigen Produkten in einer mit der Zweckbestimmung der Medizin- oder sonstigen Produkte vereinbaren Weise und innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen kombinieren, um sie in Form eines Systems oder einer Behandlungseinheit in Verkehr zu bringen, müssen eine Erklärung abgeben:
1. sonstige Produkte mit CE-Kennzeichnung;
2. In-vitro-Diagnostika mit CE-Kennzeichnung gemäß der Verordnung (EU) 2017/746;
3. sonstige Produkte, die den für sie geltenden Rechtsvorschriften entsprechen, nur dann, wenn sie im Rahmen eines medizinischen Verfahrens verwendet werden oder wenn ihr Vorhandensein im System oder in der Behandlungseinheit anderweitig gerechtfertigt ist.
MDR Artikel 22, Absatz 1c
Der Begriff „sonstige Produkte“ ist in 1. und 3. unterschiedlich zu verstehen. Bei 1. ist ein Produkt gemäß Artikel 1, Absatz 4 ein Medizinprodukt, ein Zubehör oder ein Produkt gemäß Anhang XVI, das den Anforderungen der MDR genügen muss.
In 3. ist mit „Produkt“ ein Produkt im umgangssprachlichen Sinn gemeint, das nicht den Anforderungen der MDR unterliegt, aber konform mit anderen regulatorischen Anforderungen sein muss und daher ggf. auch eine CE-Kennzeichnung trägt.
Damit können Medizinprodukte und explizit auch Nicht-Medizinprodukte als System bzw. einer Behandlungseinheit in Verkehr gebracht werden.
Die Nicht-Medizinprodukte müssen die geltenden (europäischen) Rechtsvorschriften einhalten. Beispiele für solche Rechtsvorschriften sind EU-Verordnungen und EU-Richtlinien wie REACH (1907/2006/EG) und die Funkanlagen-Richtlinie (2014/53/EU).
Artikel 22 „Systeme und Behandlungseinheiten“ Absatz (2)
Absatz (2) fordert ebenfalls die Erklärung, die nun etwas genauer ausfallen und mit denen der Hersteller bestätigen muss, dass er die notwendigen Informationen bereitgestellt und eine geeignete Überwachung, Überprüfung und Validierung vorgenommen hat.
In der Erklärung gemäß Absatz 1 gibt die betreffende natürliche oder juristische Person an, dass sie
1. die gegenseitige Vereinbarkeit der Medizin- und gegebenenfalls sonstigen Produkte entsprechend den Hinweisen der Hersteller geprüft und ihre Tätigkeiten entsprechend diesen Hinweisen durchgeführt hat,
2. das System oder die Behandlungseinheit verpackt und die einschlägigen Benutzerhinweise angegeben hat, unter Einbeziehung der Informationen, die vom Hersteller der Medizin- und sonstigen zusammengestellten Produkte bereitzustellen sind,
3. die Zusammenstellung von Medizin- und gegebenenfalls sonstigen Produkten zu einem System oder einer Behandlungseinheit unter Anwendung geeigneter Methoden der internen Überwachung, Überprüfung und Validierung vorgenommen hat.
Die Forderung der MDR in diesem Artikel 22 Absatz 2 (3) nach „Überprüfung und Validierung“ geht über die entsprechende Anforderung der MDD in Artikel 12 (2)(c) hinaus. Demnach muss der Hersteller „nur“ bestätigen, dass „die gesamte Tätigkeit in geeigneter Weise intern überwacht und kontrolliert wurde.“
Überprüfung und Validierung (im Englischen „Verification and Validation“) erfordern implizit Vorgaben, gegen die der Hersteller verifizieren und validieren kann. Das wiederum bedingt beispielsweise eine Spezifikation des „kombinierten“ Systems. Eine solche (indirekte) Anforderung enthält die MDD nicht.
Artikel 22 „Systeme und Behandlungseinheiten“ Absatz (4)
Absatz (4) erwähnt erneut den Begriff „Produkte“:
Enthält das System oder die Behandlungseinheit Produkte, die keine CE-Kennzeichnung tragen, oder ist die gewählte Kombination von Produkten nicht mit deren ursprünglicher Zweckbestimmung vereinbar oder wurde die Sterilisation nicht gemäß den Anweisungen des Herstellers durchgeführt, so wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen. Die natürliche oder juristische Person ist den für die Hersteller geltenden Pflichten unterworfen.
Im englischen Text heißt es:
Where the system or procedure pack incorporates devices which do not bear the CE marking
Es sind somit die Produkte in der Definition des Artikels 1, Absatz 4 gemeint.
Die MDD verlangte eine erneute Konformitätsbewertung bei Systemen oder Behandlungseinheiten, wenn diese umfassen:
- Medizinprodukte, die eine CE-Kennzeichnung tragen, sowie
- Nicht-Medizinprodukte oder/und
- Medizinprodukte, die nicht in ihrer ursprünglichen Zweckbestimmung eingesetzt werden
Hingegen fordert die MDR eine erneute Konformitätsbewertung bei Systemen oder Behandlungseinheiten, wenn diese umfassen:
- Medizinprodukte, die eine CE-Kennzeichnung tragen, sowie
- Medizinprodukte, die keine CE-Kennzeichnung tragen (das könnten z. B. FDA-zugelassene Medizinprodukte sein) oder/und
- Medizinprodukte, die nicht in ihrer ursprünglichen Zweckbestimmung eingesetzt werden
Die Kombination eines Medizinprodukts mit einem Nicht-Medizinprodukt ohne erneute Konformitätsbewertung schließt die MDR im Gegensatz zur MDD wie oben dargestellt gerade nicht mehr aus.
Artikel 29 „Registrierung von Produkten“
Eine im Vergleich zur MDD zusätzliche Anforderung der MDR besteht darin, dass die Hersteller für das System bzw. die Behandlungseinheit eine UDI vergeben und in die EUDAMED eingeben:
(2) Bevor ein System oder eine Behandlungseinheit gemäß Artikel 22 Absätze 1 und 3, bei dem bzw. der es sich nicht um eine Sonderanfertigung handelt, in Verkehr gebracht wird, vergibt die zuständige natürliche oder juristische Person für das System oder die Behandlungseinheit im Einklang mit den Vorschriften der Zuteilungsstelle eine Basis-UDI-DI und gibt sie zusammen mit den anderen in Anhang VI Teil B aufgeführten zentralen Datenelementen zu diesem System oder dieser Behandlungseinheit in die UDI-Datenbank ein.
Der Anhang VI beschreibt diese Anforderungen noch genauer.
UDI: Anhang VI Teil C
Die MDR legt in Absatz 3.7 der Anhangs VI zur Unique Device Identification (UDI) fest: „Systeme und Behandlungseinheiten gemäß Artikel 22 erhalten und tragen ihre eigene UDI.“ In 6.3.1 konkretisiert sie:
„Die in Artikel 22 in Bezug genommene natürliche und juristische Person ist verantwortlich für die Kennzeichnung des Systems oder der Behandlungseinheit mit einer UDI, in der sowohl die UDI-DI als auch die UDI-PI enthalten sind.“
Schließlich regelt die MDR noch, wo die UDI aufzubringen ist:
6.3.2. Produktinhalte von Systemen oder Behandlungseinheiten tragen einen UDI-Träger auf ihrer Verpackung oder auf dem Produkt selbst.
Ausnahmen:
1. Bei Einwegprodukten für den einmaligen individuellen Gebrauch, deren Anwendung den Personen, die sie verwenden sollen, allgemein bekannt ist, die in einem System oder einer Behandlungseinheit enthalten sind und die nicht für die individuelle Verwendung außerhalb des Systems oder der Behandlungseinheit vorgesehen sind, ist es nicht erforderlich, dass sie einen eigenen UDI-Träger tragen.
2. Bei Produkten, die von der Pflicht des Anbringens eines UDI-Trägers auf der einschlägigen Verpackungsebene ausgenommen sind, ist es nicht erforderlich, dass sie einen UDI-Träger tragen, wenn sie in ein System oder eine Behandlungseinheit einbezogen sind.
6.3.3. Anbringen des UDI-Trägers auf Systeme oder Behandlungseinheiten
1. In der Regel wird der UDI-Träger eines Systems oder einer Behandlungseinheit auf der Außenseite der Verpackung angebracht.
2. Der UDI-Träger ist lesbar oder lässt sich im Falle von AIDC scannen, unabhängig davon, ob er sich auf der Außenseite der Verpackung des Systems oder der Behandlungseinheit oder innerhalb einer durchsichtigen Verpackung befindet.
MDR, Anhang VI, Absatz 6.3.2
Zusammenfassung der Änderungen durch die MDR
Die wesentlichen Änderungen, die die MDR im Gegensatz zur MDD einführt, sind:
- Systeme und Behandlungseinheiten dürfen Nicht-Medizinprodukte beinhalten.
- Die Hersteller müssen bestätigen, dass sie diese „Kombination“ verifiziert und validiert haben, was wiederum u. a. Spezifikationen bedingt, gegen die verifiziert und validiert werden kann.
- Systeme und Behandlungseinheiten bedürfen einer eigenen UDI.
f) Vorgaben der FDA an Systeme und Behandlungseinheiten
Die FDA kennt noch den Begriff des „Convenience Kits“:
„Convenience kit means two or more different medical devices packaged together for the convenience of the user.“
Quelle: FDA 21 CFR 801.3
Dieser entspricht zwar eher Behandlungseinheit, wobei die Behandlungseinheit nicht unterscheidet, ob das gemeinsame Verpacken der „Convenience“ dient. Bei dem Convenience Kit geht es auch nicht darum, dass die Produkte notwendigerweise (alle) zusammen einem medizinischen Zweck dienen.
Beispiele für Convenience Kits sind Erste-Hilfe-Kästen oder Sets von Produkten, die für eine bestimmte Operation bereits zusammen verpackt wurden. Die Anforderungen an die Vergabe von UDI an diese Kits hat die FDA in einem eigenen Guidance Document „UDI Convenience Kits“ publiziert.
Die FDA kennt zudem noch „Device Packages“:
„Device package means a package that contains a fixed quantity of a particular version or model of a device.“
Quelle: FDA 21 CFR 801.3
Dies entspricht aber nicht den Systemen und Behandlungseinheiten im Sinne der europäischen Gesetzgebung.
3. Zusammenfassung & Fazit
a) Regulatorische Anforderungen
Die MDR hat im Vergleich zur MDD dahingehend eine Erleichterung eingeführt, dass Systeme und Behandlungseinheiten Nicht-Medizinprodukte enthalten dürfen. Diese müssen die Hersteller gemäß MDR verifizieren und validieren, was die MDD so nicht explizit forderte.
Die mangelnde Übersetzung der MDR ins Deutsche führt zu missverständlichen und irreführenden Anforderungen, insbesondere im Artikel 22.
Die MDR bedarf im Gegensatz zur MDD keiner Überführung in nationales Recht mehr. Bei der Frage, wie gut diese bei der MDD gelang, kommt RA Dr. Banz zum Schluss:
„[…] ist es nahezu unmöglich, die bei wörtlicher Anwendung von §10 Abs. 2 MPG entstehenden Hürden und Anforderungen zu erfüllen. Diese Schwierigkeiten, verbunden mit dem Fehlen jeder Begründung des Gesetzgebers für das Überschreiten des durch Art. 12 Abs. 2 S. 2 MDD vorgegebenen Rahmens, führen zu dem Ergebnis, dass möglicherweise doch eine unsaubere Implementierung der Richtlinie in nationales Recht vorliegt.“
Den Artikel „Systeme und Behandlungseinheiten und ihre Behandlung nach dem Medizinprodukterecht“ von Banz/Eckle/Kremser finden Sie online sowie in MPR2/2009 S. 50 ff.
Die Tatsache, dass sich die Behörden selbst uneins darüber sind, wann eine Erklärung zu einem System oder einer Behandlungseinheit abgegeben werden muss (s. o.), schafft weitere Rechtsunsicherheit.
b) Unkenntnis der regulatorischen Anforderungen
Vielen Herstellern sind die regulatorischen Anforderungen nicht bekannt, die sie erfüllen müssen, wenn sie Systeme und Behandlungseinheiten verkaufen wollen. Armin Gärtner schreibt uns dazu:
„Die Aufsichtsbehörden verlangen zunehmend in Krankenhäusern die Artikel–12-Erklärungen, z. B. für Gerätewagen der Endoskopie, aber auch für andere Systeme, die von einem Hersteller geliefert wurden. Leider machen das viele Hersteller aus Unkenntnis nicht. Andere verschieben die Verantwortung für das Zusammenstellen von verkauften Einzelprodukten auf die Krankenhäuser, die das in der Regel nicht wissen und nicht verstehen.“
c) Abgrenzungen
Dieser Artikel hat nicht die Kombinationsprodukte (Medizinprodukte und Arzneimittel) diskutiert. Er ist auch nicht darauf eingegangen, wann es sinnvoll ist, Produkte einzeln, als Systeme oder Behandlungseinheiten oder als ein Produkt zu vermarkten.
Eine Diskussion der Vor- und Nachteile beim Kombinieren von Medizinprodukten finden Sie in einem separaten Artikel.
Update: Eine ältere Version dieses Artikels bezog sich auf die fehlerhafte deutsche Übersetzung der MDR und kam daher zu inkorrekten Schlussfolgerungen. Diese wurden behoben. Danke an Jörg Schmidt.
Änderungshistorie
- 2024-08-06: Verweis in 1.d) ergänzt. Kapitel 2.a) bis 2.d) in Vergangenheitsform umformuliert
- 2020-03-18: Erste Version des Artikels



Sehr informativer Artikel. Ggf. wäre es interessant in einer zukünftigen Revision noch den Systembegriff der IEC 60601-1 gegenüberzustellen.
Sie haben absolut Recht, lieber Herr Müller! Das sollte und werde ich ergänzen!
Danke für den Hinweis!
Beste Grüße
Christian Johner
Sehr geehrtes Johner Team,
folgende Frage hätte ich. Wenn ein medical PC dafür eingesetzt wird, dass aus analogen Bilder von MG in digitale umgewandelt wird. Wie würden Sie den PC einstufen?
Lieber Herr Müller,
Vielen Dank für Ihre Frage. Ich bin mir noch unsicher, in Bezug auf welche Überlegung Sie den PC einstufen möchten. Der Begriff „Medical PC“ bezieht sich in der Regel auf das Thema der elektrischen Sicherheit. Wenn der PC eine Verbindung zu einem Medizinprodukt hat, möglicherweise einem Röntgengerät, und es über diese galvanische Verbindung, meistens über den Schirm, zu einem erhöhten Patientenableitstrom am Medizinprodukt kommen kann, müsste eine zusätzliche Trennung eingebaut werden. Ein Medical PC erfüllt oft nicht die Ableitstrombedingungen für den Patientenschutz, selbst wenn ein Netzteil nach IEC 60601-1 verbaut wurde. Das Kapitel 16 der IEC 60601-1 nennt die Anforderungen für ME-Systeme. Wenn der PC keine solchen kritischen Verbindungen hat und außerhalb der Patientenumgebung betrieben wird, können Sie einen handelsüblichen PC verwenden. Der PC muss natürlich die Anforderungen an Rechenleistung und Speicherkapazität aus Sicht der Software erfüllen. Das wäre dann eine Anforderung gemäß IEC 62304. Sie benötigen jedoch keinen speziellen Medical PC dafür.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck,
viele Medizinproduktgeräte erstellen noch analoge Objekte. Mithilfe eines Medical PC’s (galvanische Trennung, EMV, etc. wird erfüllt) und einer speziellen Software, werden diese Bilder digitalisiert und an ein Medizinprodukt gesendet. Wird nun der Medical PC an die Medizingeräte angeschlossen, fordern die Techniker Messung der Ableitströme und sonstiges. Wie kann man am besten den Medical PC platzieren, sodass die Techniker zufrieden sind und die Bilder digitalisiert werden. Eine Option wäre den Medical PC und die Software zu einem Medizinprodukt zu machen. Dies wäre ein enormer Aufwand. Hätten Sie eine andere Idee?
Lieber Herr Müller,
Vielen Dank für Ihre Frage!
Ich verstehe Ihre Beschreibung so, dass Ihr PC als analog-digital Converter (System A) zwischengeschaltet ist, zwischen dem bildgebenden Gerät (System B) und einem anderen Medizinprodukt (System C). Wenn das System (C) keine Verbindung zu einem Patienten hat, müssen Sie zwischen (A) und (C) nichts weiter beachten. Wenn System (B) eine Verbindung zum Patienten hat, dann müssen Sie zwischen (A) und (B) elektrisch sicher sein. Diese Sicherheit (1MOPP@230V), kann bereits am Signalausgang von (B) integriert sein, dann müssen Sie nichts machen. Falls Sie es nicht wissen, müssten Sie die Sicherheit zur Verfügung stellen. Der Aufstellort würde nur eine Rolle spielen, wenn es eine Verbindung vom Patienten zu Ihrem System (A) geben kann.
Die Trennung zwischen (A) und (B) erreichen sie durch folgende Möglichkeiten
– eine galvanische Trennung im Verbindungskabel A-B
– Schirm vom Verbindungskabel nicht auflegen (nicht optimal)
– Medical PC mit Trennung im Netzteil + Messen der Ableitsröme am Ausgang zu (B) (Die Techniker haben recht)
Die IEC 60601-1 hat im Anhang I in der Tabelle Table I.1 eine Übersicht aller Konstellationen einer ME-Systembildung und dort finden Sie auch die möglichen Massnahmen.
Herzliche Grüsse, Mario Klessascheck
Sehr geehrter Herr Prof. Johner, wir packen zu unserem langzeit-verwendbaren Medizinprodukt der Klasse 2 b weitere ce-gekennzeichnete Medizinprodukte anderer Hersteller hinzu, ohne sie der Originalverpackung zu entnehmen (disposables: Skalpell, Steriltuch, Einweghandschuhe, Luer-Hähne). Gemäß unserer anliegenden Gebrauchsinformation benötigt der Anwender die Teile zum „final processing“ unserer Produkte, d.h. zum aseptischen Öffnen und Vorspülen am Tag vor der Behandlung. Zwar lassen die Anwender die Hähne dann in der Regel am Produkt, die Entscheidung liegt aber beim Anwender, d.h. er kann dann zur 1. Behandlung auch eigene, neue Hähne verwenden. Nach unserem Verständnis handelt es sich somit nicht um ein „procedure pack“ und Art. 12 ist nicht anwendbar, da die genannten Artikel überhaupt nicht bei der Behandlung verwendet werden, sondern Werkzeuge zur Installation sind. Unsere Benannte Stelle hat uns wegen Fehlens der Art. 12-Erklärung eine major-nonconformity reingewürgt. Ist das berechtigt? Da uns im Zusammenhang mit anderen merkwürdigen Sichtweisen das Zertifikat suspendiert wurde, würde ich mich sehr freuen, wenn Sie mir Ihre Sichtweise zu diesem Problem mitteilen würden. Mit freundlichen Grüßen, Karsten Thies
Sehr geehrter Herr Dr. Thies,
danke für diese sehr spannende Frage. Wenn Sie die Produkte, die eigenen und die anderer Hersteller, gemeinsam verpacken und in den Verkehr bringen, liegt meines Erachtens schon eine Behandlungseinheit vor. Daran ändert weder die Tatsache, dass die Anwender nicht immer alle Produkte dieser „Einheit“ nutzen, noch dass diese Produkte nur der Installation dienen. Bei Produkten für eine Installation muss man sich aber fragen, ob ein Medizinprodukt oder eher ein Zubehör vorliegt. Für Zubehör gelten „dummerweise“ nur die gleichen Anforderungen wie für die Medizinprodukte selbst. Und genau diesem Sachverhalt scheinen Sie zum Opfer gefallen zu sein.
Eine Major-Non-Conformity ist ärgerlich, und das tut mir leid. Sie ist zum Glück eine schnell zu behebende Abweichung. Weshalb Ihnen das Zertifikat suspendiert wurde, kann ich nicht nachvollziehen. Das ist ja wirklich ärgerlich. Geben Sie einfach Bescheid, wenn wir helfen können.
Viele Grüße
Christian Johner
Sehr geehrter Prof. Johner,
Nachdem ich mir den Artikel in letzter Zeit mehrfach durchgelesen habe, will mir immer noch nicht ganz eingehen, wo das System aufhört und wie weit die Systemerklärung tatsächlich greifen muss.
Es geht ja nur um Produkte, die ich im Rahmen einer medizinischen Zweckbestimmung kombiniere.
Habe ich also beispielsweise eine Software, die im Rahmen der medizinischen Zweckbestimmung Biosignal-Daten von einem Aufnahmegerät erhält und anzeigt, ist das eine System im Sinne von Art. 12, richtig? Sowohl die Software, als auch das Aufnahmegerät sind Medizinprodukte, tragen ein CE-Zeichen und sind dazu bestimmt, miteinander verwendet zu werden.
Ist die Software dazu bestimmt, auf einem beliebigen handelsüblichen PC verwendet zu werden, der die ausgewiesenen Mindestanforderungen erfüllt, gehört dieser aber nicht mehr zu diesem System dazu.
Gleiches würde also in weiterer Folge für Gerätewagen, Trenntrafo und PC-Peripherie gelten, da diese ja alle keine medizinische Zweckbestimmung erfüllen, oder?
Alles zusammen müsste also keine gemeinsame Systemerklärung gemäß Artikel 12 besitzen.
Anders sieht die Sache jedoch in Bezug auf ein ME-System gemäß IEC 60601 aus. Hier zählt der gesamte Wagen samt aller Verkabelung zu einem ME-System.
Was ist nun hier die korrekte Vorgehensweise?
Herzlichen Dank!
Hallo,
die Artikel 12 Erklärung gilt für Hersteller (bzw. Fachhändler), die Systeme aus Medizinprodukten erstellen. Artikel 12 enthält auch den Verweis auf Artikel 11, wenn Medizinprodukte mit Nichtmedizinprodukten zu einem System erstellt werden. Dann ist nach Artikel 11 eine neue Konformitätsbewertung fällig.
Also es gilt folgender Ansatz
Hersteller (Fachhändler) erstellt System aus Medizinprodukten = Artikel 12 Erklärung
Hersteller (Fachhändler) erstellt System aus Medizinprodukten + Nichtmedizinprodukten = Artikel 11 neues Konformitätsbewertungsverfahren
(Hinweis: Letzteres wird in der Praxis häufig nicht gemacht)
Betreiber
Betreiber stellt Medizinprodukte mit Medizinprodukten, Zubehör, Software und andere Gegenstände wie ein Endoskopiewagen zu einer neuen Kombination unter Berücksichtigung der Zweckbestimmung(en) zusammen. Dann greift § 4 Abs. 4 der Medizinprodukte-Betreiberverordnung. Das gilt auch für den Fall, dass der Betreiber eine Herstellerfirma oder Fachhändler oder Serviceorganisation beauftragt.
§ 4 ABs. 4 verlangt weiter, dass der Betreiber die Kombination nur erstellen darf, wenn diese für die Sicherheit der Patienten, Anwender, Beschäftigten und Dritten geeignet ist. Diesen Nachweis führt man mit einer Technischen Dokumentation incls. Risikoanalyse. Dazu verwendet man auch die Anforderungen von Kapitel 16 ME-Systeme der DIN EN 60601-1.
DIeses Verfahren hat sich bewährt und wird auch von Aufsichtsbehörden akzeptiert.
Solange ein Betreiber bei einer solchen Kombination (auch bei Änderungen) im Rahmen der Herstellerzweckbestimmungen nachweislich bleibt, greift auch nicht das ominöse Thema Eigenherstellung § 12 MPG. Das Thema Eigenherstellung ist aus unbekannten Gründen in der Betreiberverordnung nicht ausgeführt; eigentlich sollte man das in einer Ausführungsverordnung erwarten. Auch die EU-Medizinprodukteverordnung enthält zwar einen Passus zur Eigenherstellung, er ist aber aus praktischer SIcht unklar und ohne präzise Angaben/Vorgaben formuliert…..
Leider hat der Verordnungsgeber (Bundesministerium für Gesundheit i Berlin) es bis heute nicht geschafft, die sehr abstrakte und technikneutrale Formulierung des § 4 Abs. 4 in der nationalen MPBetreibV transparent und eindeutig praxisgerecht zu formulieren.
mfg
Armin Gärtner
Einmal mehr herzlichen Dank, lieber Herr Gärtner!
Ihre Ergänzungen schätze ich sehr wert!
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ist Ihnen bekannt, ob nach der MDD ausgestellte Artikel-12-Erklärungen auch noch über den 25.05.2020 gültig bleiben? Z.B. in Analogie zur CE-Erklärung bis 4 Jahre nachreichend? Oder ist nach Ihrer Auffassung/ Kenntnis ab dem 26.05.2020 zwingend eine Artikel-22-Erklärung gemäß MDR erforderlich – auch für davor in Verkehr gebrachte Systeme?
Besten Dank vorab für Ihre Beantwortung.
Andreas Keller
Sehr geehrter Herr Keller,
danke für Ihre spannende Frage, auf die es meines Wissens noch eine definitive Antwort gibt. Das hängt auch damit zusammen, dass die nationalen Gesetze und Verordnungen, die die Details regeln nicht an die MDR angepasst sind und wahrscheinlich auch nicht mehr rechtzeitig fertig werden.
Die MDR enthält keine spezielle Übergangsfrist für diese „Kombinationen“. Daher würde ich davon ausgehen, dass die Erklärung solange gültig ist, solange alle(!) Produkte dieses Systems bzw. dieser Behandlungseinheit legal in Verkehr gebracht werden dürfen. D.h. die Übergangsfristen könnten die Gültigkeit über den 25.05.2020 verlängern.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich würde gerne ihre Interpretation zu folgendem Thema hören:
Ist es zwingend notwendig, als Inverkehrbringer eines Systems / Behandlungseinheit, aus mehreren CE gekennzeichneten Medizinprodukten (Anwendung innerhalb der jeweiligen Zweckbestimmung) eine QSV mit dem Herstellern der jeweiligen Produkte abzuschließen?
Und, benötigtigen wir weitere Dokumente vom den Herstellern außer der Konformitätserklärung und der Gebrauchsanleitung insofern wir die gegenseitige Vereinbarkeit der Produkte verifiziert haben?
Viele Grüße
Fabian Kaiser
Sehr geehrter Herr Kaiser,
Sie brauchen nicht ganz zwingend eine QSV. Aber es wäre hilfreich. Sonst ist es schwieriger sicherstellen, dass Sie informiert werden, wenn es mit dem jeweiligen Produkt Schwierigkeiten oder Änderungen gibt.
Die beiden genannten Dokumente reichen. Die Zweckbestimmung wäre ggf. noch hilfreich, falls die Gebrauchsanweisung nicht ausreicht.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich habe eine Frage bzgl. dieser Thematik.
Sehe ich das richtig, dass man als Ersteller einer Behandlungseinheit (sofern Artikel 22 zutrifft) kein Hersteller (im Rahmen der MDR) ist und somit nicht zwingend ein Qualitätsmanagementsystem (zB nach ISO13485) benötigt?
Sehr geehrter Herr Gostner,
Danke für Ihre spannende Frage!
Die Frage lässt sich nicht generell bejahen. Gerade die Abschnitte 3. und 4. nennen Fälle, in denen die „Firma“ doch zum Hersteller wird bzw. doch ein QM-System benötigt.
Wenn jemand jedoch nicht-sterile Produkte als Behandlungseinheit zusammen stellt, wobei jedes der Produkte im Rahmen der vorgesehenen Zweckbestimmung eingesetzt wird, benötigt dieser nicht zwingend ein (zertifiziertes) QM-System.
Die deutsche Version der MDR ist auch im Artikel 22 nicht präzise. Bitte verwenden Sie immer die englische Version.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen herzlichen Dank für Ihre schnelle, wertvolle Antwort und für Ihren Tipp mit der englischen Version der MDR!
Viele Grüße,
Daniel Gostner
(PS: Die Verabschiedung habe ich in meinem ersten Kommentar unhöflicherweise vergessen da ich zu schnell auf absenden geklickt habe)
Sehr geehrter Herr Professor Johner,
ich hätte eine generelle Frage zu Sets, im konkreten Fall bestehend aus CE-gekennzeichneten Medizinprodukten (Blasenkatheter und Blockermedium, steril verpackt) und einem Nicht-CE gekennzeichnete Arzneimittel, das zur Unterstützung verwendet wird (Gleitgel). Gelten Arzneimittel als sonstige Produkte und muss daher die MDD angewandt werden, oder liegt ein solches Set ausserhalb des Geltungsbereiches der MDD/MDR?
Sehr geehrter Herr Gebhardt,
die Antwort auf Ihre Frage hängt wirklich davon ab, ob wir von MDR oder MDD sprechen. Geben Sie mir kurz ein Zeichen, was wichtiger ist. Die Antwort ist kein Einzeiler.
Danke für Ihre Unterstützung!
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Nachricht.
es betrifft Produkte, für die wir die Übergangsfrist nach Art.120 (MDR) in Anspruch nehmen und weiterhin das gültige RL-Zertifikat verwenden.
Gruß
Thomas Gebhardt
Sehr geehrter Herr Professor Johner,
inwiefern gibt es zwischen MDD und MDR einen Unterschied, wenn ich CE-gekennzeichnete Produkte mit nicht CE-gekennzeichneten Produkten (in unserem Fall ein nicht zulassungspflichtiges Arzneimittel) in einer Verpackungseinheit in Verkehr bringen möchte?
Wenn ich die MDCG 2018-3 Guidance richtig verstanden habe, handelt es sich nur dann nicht um ein System/Behandlungseinheit, wenn ich die Einzelkomponenten nicht auch getrennt in Verkehr bringe, oder? Wenn es also kein System/Behandlungseinheit wäre, als was wäre es dann einzustufen?
Dazu würde mich Ihre Meinung sehr interessieren.
Gruß
Thomas Gebhardt
Sehr geehrter Herr Gebhardt,
wenn Sie Medizinprodukte zusammen mit einem Arzneimittel gemeinsam in Verkehr bringen, dann ist das kein System im Sinne der MDR. Es handelt sich um Kombinationsprodukt. Hierfür gelten weitere regulatorische Anforderungen u.a. von der Arzneimittelrichtlinie.
Hilft das schon ein wenig? Ich plane demnächst einen Beitrag zu Kombinationsprodukten.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
vielen Dank für Ihre Einschätzung.
Das wirft ein ganz anderes Licht auf den Vorgang, würde es sich auch dann um ein Kombinationsprodukt handeln, wenn die arzneimittelhaltigen Komponenten des Sets ausschliesslich als Hilfsmittel für die Behandlung verwendet werden (Desinfektionsmittel, Gleitmittel)?
Einen Beitrag zu Kombinationsprodukten würde ich sehr begrüßen.
Gruß
Thomas Gebhardt
Sehr geehrter Herr Gebhardt,
wenn es sich nur um ein „Set“ handelt, dann ist es kein Kombinationsprodukt. Ein Kombinationsprodukt ist ein Produkt, das beides „kombiniert“. Ob diese Form eines Sets als System gemäß MDR Artikel 22 Absatz 1c Spiegelstrich 3 verstanden werden kann, müssten wir im Einzelfall prüfen.
Viele Grüße, Christian Johner
Sehr geehrter Prof. Johner
Vielleicht müsste man die MDCG 2018-3 Guidance hier ergänzen, da dort eine Einordnung vorgenommen wird, wann es sich nicht um eine Behandlungseinheit handelt.
Viele Grüsse,
Christian Weckert
Danke für den Hinweis, lieber Herr Weckert!
Ich werde das zeitnah ergänzen.
Herzliche Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihren informativen Artikel zum Thema System- und Behandlungseinheiten.
Mein Unternehmen bietet Austauschsets für z.B. Sauerstoffkonzentratoren an. In diesen Sets befinden sich Zubehörteile und Ersatzteile wie Nasenbrillen, Filter, Anschlusstüllen, etc.
Handelt es sich bei solch einer Zusammenstellung an verschiedenen Produkten, in Ihren Augen um eine System-oder Behandlungseinheit? Würde es ggf. einen Unterschied machen, wenn diese auf Kundenwunsch zusammen gestellt werden?
Vielen Dank im Voraus für Ihre Meinung!
Schöne Grüße
M. Korf
Sehr geehrte/r M.Korf,
danke für Ihre Frage!
Sie schreiben, dass sich in den Sets „Zubehörteile“ befinden würden. Ich bin aber unsicher, ob Sie damit Zubehör im Sinne der MDR bzw. MDD meinen. So wie ich den Rest der Frage verstehe, sind das eher Teile — Ersatzteile — für Medizinprodukte.
Wenn diese Vermutung korrekt ist, sind Ihre Sets keine Systeme- oder Behandlungseinheiten. Denn diese müssen Medizinprodukte enthalten. Ein Set, dass „nur“ Austauschteile, aber kein CE-kennzeichneten Medizinprodukte oder Zubehöre enthält, fällt nicht unter die Definition.
Mit besten Grüßen, Christian Johner
Sehr geehrte Herr Johner,
wie sehen sie die Sache mit Nachbausensoren (z.B. Pulsoxymetrie) die wir auf den Stationen verwenden die aber in keiner Gebrauchsanleitung explizit vom Orginalhersteller angeführt sind? Verstößt man hier nicht gegen die gültige ISO-Norm wie die 80601-2-61? Es wundert mich auch das wir hier eine Kompatibilitätsbescheinigung bekommen aber hier keine Testung mit den Endgeräten durchgeführt wurde. Ist sowas überhaupt rechtens?
Vielen Dank für Ihr Feedback.
Sehr geehrter Herr Pisac,
danke für Ihre heutige Frage!
Ich bin nicht ganz sicher, ob ich alles verstehe. Falls ich also an der Sache „vorbei-antworten“ sollte, haken Sie bitte nach.
Wenn eine Firma Sensoren (ich vermute, damit ist nicht nur der Sensor im Sinne eines Bauteils, sondern ein Produkt gemeint) in den Verkehr bringt, dann ist dieser Sensor mit höchster Wahrscheinlichkeit als Zubehör zu qualifizieren.
Dieses Zubehör muss alle regulatorischen Anforderungen z.B. der MDR bzw. MDD erfüllen. Dazu zählen die grundlegenden Anforderungen. Zum Nachweis dieser Anforderungen sind Hersteller angehalten, harmonisierte Normen zu befolgen. Nach meinem Wissen zählt die EN 80601-2-61 im Gegensatz zu anderen Normen der Serie EN 80601-2 nicht dazu (Quelle. Dennoch dürfte auch die genannte Norm den Stand der Technik repräsentieren. Ob der Hersteller diese Norm einhält, kann ich nicht beurteilen.
Einen zweiten Punkt, den Sie ansprechen, ist der bestimmungsgemäße Gebrauch. Wenn der Hersteller des „Endgeräts“ sich dazu nicht auslässt bzw. den Gebrauch anderer als der eigenen Sensoren nicht untersagt, dann kann der Sensor-Hersteller durchaus solch ein Zubehör in den Markt bringen. Wie gesagt muss sein Produkt die grundlegenden Anforderungen erfüllen, wozu auch Kompatibiltätsanforderungen zählen. Ob der Sensor-Hersteller die Einhaltung all dieser Anforderungen geprüft hat, kann ich nicht beurteilen.
Der dritte Punkt betrifft das Thema des Beitrags, den Sie kommentieren, nämlich die Systeme und Behandlungseinheiten. Falls der Sensor-Hersteller nur seinen Sensor d.h. ohne das „Endgerät“ in den Verkehr bringt, dann liegt kein System bzw. keine Behandlungseinheit z.B. im Sinne der MDR Artikel 22 (MDD analog) vor.
Beste Grüße, Christian Johner
Guten Morgen Prof. Johner,
vielen Dank, Ihre Erklärung ist sehr hilfreich.
Ich versuch nur noch für mich es besser zu verstehen und möchte es kurz erläutern:
Auf der Intensivstation haben wir z.b. die Multiparametergeräte von einem großen namhaften Hersteller. Dieser Hersteller verweist in seiner Gebrauchsanleitung auf die ISO-Norm 80601-2-61 welche Sensoren validiert wurden, sowohl die er anbietet aber auch von anderen Herstellern wo die Bezeichnung explizit angeführt ist (incl. Toleranzabweichungen usw.)
Im Rahmen von Kosteneinsparungen stellt der Einkauf dann auf billigere Nachbausensoren von Herstellern die nicht im Manual angeführt sind. Auf Nachfrage bei diesen Herstellern bekommt man meist nur eine Kompatibilitätserklärung.
Und hier stellt sich für mich die Frage ob das gesetzeskonform ist, meiner Meinung nicht da der Orginalhersteller des ME-Systems diese Sensoren nicht validiert hat und wir uns an das Manual halten müßen, oder?
Vielen Dank für Ihr Feedback.
Sehr geehrter Herr Pisac,
wenn ich sie richtig verstehe, gibt der Hersteller des Multiparametergeräts genau an, mit welchen Sensoren seine Geräte verwendet werden sollen. Dann ist jede Verwendung mit anderen Sensoren außerhalb des sogenannten „bestimmungsgemäßen Gebrauchs“. Das wäre nicht konform. Hier scheint an der falschen Stelle gespart zu werden.
Beste Grüße, Christian Johner
Sehr geehrter Hr. Prof. Dr. Johner,
Vielen Dank für Ihre ausführliche Darstellung des Sachverhalts. Hierzu hätte ich noch eine kurze Frage:
Gehe ich richtig in der Annahme, dass ich ein Medizinprodukt mit Nicht-Medizinprodukten gemeinsam als Set auf dem Markt bereitstellen kann, und dies weder als Behandlungseinheit noch als System gewertet wird, sofern die Nicht-Medizinprodukte nicht für die Erfüllung der Zweckbestimmung des Medizinprodukts notwendig sind, sondern eine unabhängige „Zweckbestimmung“ verfolgen?
mit freundlichen Grüßen,
Erwin Cetl
Sehr geehrter Herr Cetl,
danke für Ihre spannende Frage.
In der Tat setzen Systeme und Behandlungseinheiten im Sinne MDR Artikel 2 Absatz 10 und 11 den gemeinsamen Zweck voraus.
Falls es aber keine Systeme und Behandlungseinheiten sind, entfällt damit die Möglichkeit von den Bestimmungen des Artikels 22 Gebrauch zu machen. Dann wird eine Konformitätsbewertung des „Sets“ notwendig.
Beste Grüße, Christian Johner
Sehr geehrter Hr. Prof. Dr. Johner,
Vielen Dank für Ihre rasche Antwort.
Vielleicht habe ich mich zuvor ungenau ausgedrückt, jedoch ist für mich leider nicht ersichtlich weshalb eine Konformitätsbewertung des Sets benötigt wird, basierend auf der Tatsache, dass wir aus organisatorischen Gründen mehrere Produkte zusammen verkaufen möchten.
In unserem speziellen Fall wollen wir für Schulungszwecke dem Anwender sowohl das Medizinprodukt, als auch weitere Produkte, welche weder im Zusammenhang mit dem Medizinprodukt verwenden werden noch für dessen Zweckbestimmung erforderlich sind, gemeinsam als Schulungs-Set anbieten. Unserer Überlegung nach würden wir die Nicht-Medizinprodukte (Art. 001) mit dem Medizinprodukt (Art. 002) als Schulungs-Set (Art. 003) anbieten und lediglich die Konformitätserklärung des Medizinprodukts dem Set beilegen.
Diese Überlegung basiert auf der Tatsache, dass wenn der Anwender beide Produkte (Art.001 und Art.002) separat erwirbt, ebenso keine zusätzliche Konformitätsbewertung notwendig ist.
mit freundlichen Grüßen,
Erwin Cetl
Sehr geehrter Herr Cetl,
in Ihrer zweiten Nachricht finde ich Informationen, die ich in der ersten Nachricht nicht erkannte. Diese sind aber wesentlich:
Wenn Sie ein Produkt zu Schulungszwecken in den Verkehr bringen, dann greifen die Regularien nicht. Das Produkt ist kein Medizinprodukt, weil es nicht einem der Zwecke dient, die die Definition des Begriffs Medizinprodukt beinhaltet. Sie müssten allerdings Sorge tragen, dass dies klar erkenntlich ist, dass das Produkt nicht der Diagnose, Therapie, Überwachung von Krankheiten und Verletzungen dient, sondern der Schulung.
Möglicherweise hat sich die Problemstellung damit gelöst.
Beste Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
ich hätte eine grundsätzliche Frage zum Vorgehen beim Systemen und Behandlungseinheiten und hoffe, dass ich diesen Punkt nicht einfach überlesen habe. In den Definitionen heißt es „… zusammen verpackt sind…“. Dies bedeutet, dass jedes einzelne CE gekennzeichnete Produkt in seiner ursprünglichen Verpackung verbleibt und diese in eine weitere Verpackung eingebracht werden? Im Umkehrschluss: ich darf z.B. nicht Sterilprodukte aus ihrer Handelsverpackung entnehmen und zusammen in einer neuen Handelsverpackung kombinieren?
Vielen Dank für Ihre Rückmeldung!
Beste Grüße
Nadine Langguth
Sehr geehrte Frau Langguth,
wenn Sie ein Produkt aus seiner Handelsverpackung entnehmen und neu kombinieren, könnten Sie nicht von der Erleichterungen des Artikels zu den Systemen und Behandlungseinheiten profitieren. Falls Sie das in einer Rolle als Händler täten, würden die Anforderungen des Artikel 16 greifen. Oder sie sind bereits Hersteller.
Das ergibt aus meiner Sicht auch Sinn: Die Verpackung ist etwas, was der Hersteller kontrollieren muss. Sie dient manchmal als risikominimierende Maßnahme, der Hersteller muss z.B. bei der Abschätzung von Lebensdauern und den Folgen von Lagerung und Transport diese Verpackung genau kennen. Die kann man nicht einfach ändern, ohne die rechtlichen Pflichten eines Herstellers dieses Produkts zu übernehmen.
Jetzt hoffe ich, dass ich Ihre Frage auch richtig verstanden habe. Wenn nicht, haken Sie einfach nach.
Viele Grüße, Christian Johner
Sehr geehrter Herr Professor Johner,
wir stellen für unsere Fachhändler nach Kundenwunsch „Wiedereinsatzpakete“ für Sauerstoffkonzentratoren zusammen.
Diese beinhalten CE gekennzeichnete Medizinprodukte unterschiedlicher Hersteller (Nasenbrille, Sicherheitsschlauch, Ersatzfilter für Sauerstoffkonzentratoren). Unsere Fachhändler nutzen diese Wiedereinsatzpakete, um die Geräte bei einem Patientenwechsel neu auszustatten bzw. nutzt der Servicetechniker diese zum Austausch der Verbrauchsmaterialien beim Patienten vor Ort .
Handelt es sich bei solch einer Zusammenstellung an verschiedenen Medizinprodukten, in Ihren Augen um eine System-oder Behandlungseinheit?
Vielen Dank im Voraus.
Schöne Grüße
D. Schnabel
Sehr geehrte Frau, sehr geehrter Herr Schnabel,
danke für die Frage! Das was Sie beschreiben, hört sich in der Tat nach einer Behandlungseinheit an. Denn Sie bringen, wie Sie schreiben, Medizinprodukte und Nicht-Medizinprodukte als „Paket“ in den Verkehr.
Danke auch für dieses schöne Beispiel!
Beste Grüße, Christian Johner
Guten Abend Herr Johner,
wir stellen ein Kit zusammen, welches unter anderem unsterile Medizinprodukte enthält, von denen wir der gesetzl. Hersteller sind, aber auch ein steriles Medizinprodukt, von dem wir nicht der gesetzliche Hersteller sind.
D.h. wir müssen eine entsprechende Erklärung ausstellen – gibt es dafür ev. Vorlagen wie diese Erklärung auszusehen hat?
Die Gebrauchsanweisung des Zukaufproduktes müssen wir unverändert beilegen, da wir ansonsten das Produkt verändern würden und wir somit zum Hersteller werden würden, oder?
Am Kit selber erscheinen wir alleine als gesetzlicher Hersteller, da wir das Kit zusammenstellen? Den Zulieferer von dem einen Produkt müssen wir am Kit selbst nicht anführen, richtig?
Auf der Gebrauchsanweisung, die wir für den gesamten Kit beilegen dürfen / müssen wir aber schon das CE Zeichen anbringen, oder darf das nicht sein, weil sie sich auf das gesamte System bezieht?
Würde sich etwas an den Anforderungen ändern, wenn eines der Kitbestandteile ein IVD wäre?
Entschuldigen Sie bitte die vielen Fragen, aber dieses Thema ist leider noch vollkommen neu für mich.
Vielen Dank im Voraus!
Schöne Grüße,
Karl Heinz
Sehr geehrter Karl-Heinz,
eine Vorlage für die Erklärung ist mir nicht bekannt. Es gibt keine formalen Anforderungen an dieses Dokument.
Die Gebrauchsanweisung müssen Sie in der Tat unverändert beilegen. Sie zählt als Teil des User Interfaces, das sie auch nicht ändern dürfen.
Die Hersteller der jeweiligen Produkte müssen erkenntlich sein. Sonst wäre das eine Änderung am Produkt.
Sie zählen auch nicht als Hersteller dieses Systems bzw. dieser Behandlungseinheit. Der Begriff „Zulieferer“ ist in diesem Fall etwas missverständlich.
Sie dürfen keine CE-Kennzeichnung auf das System aufbringen, es sei denn, sie haben ein entsprechendes Konformitätsbewertungsverfahren dafür durchlaufen. Die Erklärung zu den Systemen und Behandlungseinheiten ist auch keine Konformitätserklärung im Sinne des Artikels 20 der MDR.
Viele Grüße, Christian Johner
Hallo,
gibt es einen Unterschied zwischen Set/Kit und System oder Procedure Pack? Wir haben ein Produkt, ein Gel + Apllikatoren, und wir können uns nicht so richtig darauf einigen, was es nun darstellt. Jedes dieser Produkte wird oder kann nicht einzeln erworben werden, nur in Kombination als Set. Wäre es nun ein System oder ein Procedure Pack oder sogar nichts davon, sondern eher eine Set/Kit?
Danke im Voraus
Viele Grüße
V. Frank
Sehr geehrte Frau Frank,
danke für Ihre wichtige Frage!
Ich verstehe Ihr „Set“ so, dass es zwar mehrere Produkte enthält, aber diese nicht einzeln, sondern nur in diesem „Set“ erhältlich sind. Nur wollen Sie wissen, ob dieses Set als System oder Behandlungseinheit zu verstehen ist.
Die MDR definiert die Behandlungseinheit wie folgt:
Ich nutze bewusst die englische Definition, weil es im Deutschen ein paar Fehler gibt. Es geht also um „products“ nicht um „devices“.
Man könnte also Ihre Set also so ein Procedure Pack bezeichnen. Allerdings vermute ich, dass die dahinterliegende Frage lautet, ob Sie damit die Anforderungen an diese Behandlungseinheiten erfüllen müssen, konkret Artikel 22 der MDR. Diese wenden sich aber an:
D.h. die Antwort auf die Frage ist, ob mindestens eines der „products“ der Behandlungseinheit ein „device“ (mit CE-Kennzeichnung) ist. Wenn das nicht der Fall wäre, dürften Sie den Artikel 22 nicht nutzen. Damit müsste die Kombination selbst eine CE-Kennzeichnung haben d.h. das entsprechende Konformitätsbewertungsverfahren durchlaufen haben.
Viele Grüße, Christian Johner
Hallo Herr Johner,
gibt es für ein System (lt. Definition der 60601-1) ein Typenschild, das auf dem System angebracht sein muss?
Schöne Grüße aus Hamburg,
Björn Weggut
Sehr geehrter Herr Weggut,
danke, dass Sie nachfragen. Denn damit geben Sie mir die Möglichkeit, auf ein häufiges Missverständnis hinzuweisen:
Die 60601-1 adressiert u.a. programmierbare medizinisch elektrische Systeme (PEMS). Diese Systeme sind aber nicht mit den Systemen (und Behandlungseinheiten) gemäß Artikel 22 zu verwechseln. Es gibt für letztere keine Anforderungen an ein Typenschild. Vielmehr dürften man das gar nicht aufbringen, weil man damit das Produkt verändern würde.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
vielleicht können Sie mir helfen bei dem ewigen Thema: Ist der PC Teil des medizinischen Systems oder nicht.
Wir haben ein zugelassenes Medizinprodukt, das aus einem physiologischen Verstärker inklusive Steuer- und Auswertesoftware besteht.
Damit dieses MP seinen Zweck erfüllen kann, muss es an eine PC mit der installierten SW angeschlossen werden. In der IfU finden sich die Mindestspezifikationen für einen PC.
Je nach Konfiguration steht der PC galvanisch getrennt in einem anderen Raum oder mit in der Patientenumgebung (gemeinsam an einem Sicherheitstransformator).
Von der IEC 60601-1 ist mir nach Abschnitt 16 die Einstufung als ME, non-ME und ME-System und die entsprechend notwendigen Prüfungen klar. Aber wie ist die Sachlage regulatorisch?
So wie ich es verstehe, hängt es davon ab, als was wir den PC verkaufen, ob es ein System ist oder nicht.
Wenn wir dem Kunden ein komplettes System aus PC, SW und Verstärker anbieten, dann ist das ganze ein medizinisches System und muss dementsprechend zugelassen werden. Nach MDD auf jedenfall durch Konformitätsbewertungsverfahren, da der PC kein MP ist.
Nach MDR gäbe es ja vielleicht sogar die Möglichkeit auch den PC als nicht-MP in ein Art. 22 einzubringen.
Auch wenn wir den PC „nur“ als Zubehör klassifizieren, wäre er doch zur Umsetzung des medizinischen Zwecks notwendig und damit ebenfalls einer Konformitätsprozedur unterworfen?
Wenn wir dem Kunden aber nur unseren Verstärker mit SW als MP verkaufen und zusätzlich ihm der Einfachheit halber einen passenden PC als nicht MP anbieten, müssten wir den PC nicht selbst bewerten bzw. in die Zulassung einbringen? (Dies wäre speziell für Auslandsregistrierungen sehr interssant)
Aber wenn wir es so anbieten, dann wäre doch der Kunde der Systemersteller? Der uns ggfs. beauftragt das System aufzubauen.
Bisher ging ich davon aus, dass das mitliefern eines PCs ein Art 12 (MDD) / Art.22 (MDR) System „erzwingt“, wurde aber in einer Schulung auf obige Alternative hingewiesen.
Und gilt das regulatorisch auch wenn der PC in der Patientenumgebung steht (entsprechendes 60601 Testing für Leckströme usw. vorausgesetzt)?
Spannend ist dann natürlich auch die Frage wer ggfs für die Daten und IT-Sicherheit des PCs verantwortlich ist.
Viele Grüße
Fritz Vollmer
Lieber Herr Vollmer,
Vielen Dank für Ihre Frage. Ich hoffe ich habe den Kern Ihrer Fragestellungen richtig verstanden.
Wenn Sie den PC (hat eigenes CE) mitliefern und dem Kunden damit versichern, dass die beiden Produkte (Verstärker und PC) innerhalb der von Ihnen definierten Anwendungsbeschränkung vereinbar sind, insbesondere elektrisch, müssen Sie die Vereinbarkeit auch sicherstellen und entsprechend geprüft haben. Für das System müssen Sie eine Artikel 22 Erklärung erstellen.
Anders verhält es sich, wenn Sie den PC nicht mitliefern und nur spezifizieren. Damit delegieren Sie die Verantwortung zum Nachweis der Vereinbarkeit an den Betreiber.
Für die IT-Sicherheit sind Sie und der Betreiber verantwortlich. Sie müssen die Anforderungen an das Netzwerk und an den Umgang mit Updates oder Patches definieren (siehe 60601-1 Abschnitt 14.13). Der Betreiber muss ergänzend für die Sicherheit der Daten sorgen.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Johner und Mitarbeitende,
Sie kommentieren den §22 (4) MDR mit: „Die Kombination eines Medizinprodukts mit einem Nicht-Medizinprodukt ohne erneute Konformitätsbewertung schließt die MDR […] nicht mehr aus.“
Gehe ich recht in der Annahme, dass dies nach §22 (1) c) unabhängig von CE-labels zutrifft? D.h. eine Behandlungseinheit darf Medizinprodukte/IVDs mit nicht-Medizinprodukten kombinieren, auch wenn letztere kein CE-label tragen?
Muss ich an dieser Stelle etwas beachten, wie z.B. einen Nachweis über deren Rechtfertigung innerhalb der Behandlungseinheiten zu erbringen?
Viele Grüße
Marek Deckena
Sehr geehrte Frau Deckena,
es ist möglich, im Rahmen eines Systems oder einer Behandlungseinheit auch nicht CE gekennzeichnete Produkte einzubinden. Jedoch muss in diesem Fall Artikel 22 (4) berücksichtigt werden. „Enthält das System oder die Behandlungseinheit Produkte, die keine CE-Kennzeichnung tragen […] so wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen“.
Viele Grüße
Hallo Markus,
genau das war jedoch Kern meiner Frage.
Meiner Auffassung nach trifft Art. 22 (4) MDR nicht auf „sonstige Produkte“ zu. Dem hier vorliegenden Artikel folgend liegt hier ein Übersetzungsfehler vor und Art. 22 (4) bezieht sich ausschließlich auf Medizinprodukte. Und da viele „sonstige produkte“ gar kein CE-label tragen können, frage ich mich, ob meine Auffassung korrekt ist.
VG
Marek Deckena
Lieber Herr Deckena,
der Kollege ist in wenigen Tagen zurück und wird Ihnen dann schnellstmöglich antworten.
Herzliche Grüße
Anja Segschneider | Redaktion
Hallo Herr Prof. Dr. Johner,
ich habe eine Frage zu Artikel 22, Absatz (4) „Enthält das System oder die Behandlungseinheit Produkte, die keine CE-Kennzeichnung tragen […]“. Was bedeutet „keine CE-Kennzeichnung“ hier? Geht es um Produkte, die zwar Medizinprodukte sind und keine CE-Kennzeichnung haben, oder geht es vielleicht um Produkte, die überhaupt gar keine CE-Kennzeichnung tragen (auch nicht die einer anderen für sie anwendbaren Regularie)? Wollen wir beispielsweise einen gewöhnlichen Netzstecker mit in das Procedure Pack legen, welcher aber ein CE-Zeichen trägt, gilt dann der Absatz oder nicht?
Viele Grüße
Kim Heimhuber
Liebe Frau Heimhuber,
Der Begriff Produkte im Artikel 22, Absatz 4 bezieht sich auf Medizinprodukte, Zubehör und die im Anhang XVI aufgeführten Produkte (siehe Artikel 1, Absatz 4). Damit sind Medizinprodukte aus anderen Rechtsgebieten, wie zum Beispiel der FDA gemeint. Das Beipacken des Netzsteckers steht somit nicht im Fokus dieses Absatzes.
Herzliche Grüsse, Mario Klessascheck
Sehr geehrter Herr Professor Johner,
Wir stellen aus Produkten unseres Mutterkonzerns ein Kit – im Bereich der Lymphtherapie – zusammen. Die Herstellerverantwortung für die einzelnen Produkte des Kits liegen beim Mutterkonzern.
1.Nun habe ich ein kleines Problem bei der Zuordnung System oder Behandlungseinheit?
Tendieren würde ich zur Behandlungseinheit, da die zusammen verpackten Produkte zur Reduzierung eines Lymphödems vorgesehen sind. Sicher bin ich mir aber nicht, da diese Produkte auch einzeln zu erwerben sind.
2.Welche Rolle haben wir nun inne, wenn wir dieses Kit zusammenstellen und in den Verkehr bringen?
Sind wir dann Hersteller?
3. Muss für dieses Kit ein eigenes Konformitätsbewertungsverfahren durchgeführt werden?- für den Fall, dass nicht alle enthaltenen Produkte CE-gekennzeichnet sind. Erst danach darf das Kit CE-gekennzeichnet werden?
4. Das Kit wird CE-gekennzeichnet, für den Fall, dass es ausschließlich CE-gekennzeichnete Produkte enthält.
Wahrscheinlich sind es Basics, die ich hier frage, aber leider finde ich die Definitionen nicht sehr eindeutig und bin deshalb etwas verunsichert.
Vielen Dank für Ihre Hilfe !!!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
besten Dank für Ihre Frage.
Zu 1) Wenn die Produkte nur zusammen verpackt werden, ist es eine Behandlungseinheit.
Zu 2) Wenn die Herstellerverantwortung für die einzelnen Produkte beim Mutterkonzern liegt, dann haben Sie die Rolle der Artikel 22-Person, d.h. „System/Behandlungseinheiten-Zusammensteller“
Zu 3) Wenn in der Zusammenstellung sonstige nicht-CE gekennzeichnete Produkte enthalten sind, muss die Gesamtheit als Medizinprodukt CE-gekennzeichnet werden.
Zu 4) Die Gesamtheit selbst bekommt kein eigenes CE. Außer es wird wie in #3 einem Konformitätsbewertungsverfahren unterzogen, falls sonstige nicht CE-gekennzeichnete Produkte enthalten sind.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck,
vielen Dank für Ihre Antwort, die bei mir weitere Fragen aufwirft.
Wir sind also „Zusammensteller“ des Kits.
Wie erfolgt dann die Kennzeichnung auf dem Verpackungsetikett?
Wird als Hersteller und CE-Verantwortlicher für die Bestandteile des Kits der Mutterkonzern angegeben?
Wird der Zusammensteller auch angegeben?
Das Kit besteht 100% aus CE-gekennzeichneten Produkten. Somit ist ein eigenes Konformitätsbewertungsverfahren nicht notwendig und es braucht kein eigenes CE.
Bedeutet dies, dass auf dem Etikett oder der Verpackung KEIN CE vorhanden ist? Das erscheint mir merkwürdig, da es doch für Medizinprodukte Voraussetzung fürs Inverkehrbringen ist.
Vielen Dank für Ihre Hilfe!
Mit freundlichen Grüßen
Christine Graß
Liebe Frau Graß,
meine Kollegen Herr Klessascheck und Herr Seib haben mir folgende Antwort gegeben:
Bei einem System bzw. Behandlungseinheit spricht man nicht von Inverkehrbringen, weil ja alle Komponenten für sich bereits rechtmäßig in Verkehr gebracht wurden (CE-Kennzeichnung). Artikel 22 der MDR sagt genau für solche Fälle, dass der Zusammensteller auf dem Etikett zwar auf dem Etikett genannt wird, das System aber keine eigene CE-Kennzeichnung tragen darf. Das wäre nur nötig, wenn dies als eigenständiges Produkt mit eigener Zweckbestimmung auf den Markt gebracht würde. Dann wäre der Zusammensteller auch Hersteller mit allen zugehörigen Verpflichtungen.
Zusätzlich ist noch eine Erklärung nötig, dass alle Komponenten miteinander gemäß ihrer Zweckbestimmung vereinbar sind – das gilt eben nur, wenn alle Produkte in CE-Zeichen tragen.
Herzliche Grüße
Anja Segschneider | Redaktion
Eine Frage zur Registrierung als „Zusammensteller“ nach Artikel 22 in der Eudamed-Datenbank.
Bis jetzt kann man sich nur als Hersteller, EU.Bevollmächtigter und Importeur registrieren und auch Daten eingeben.
Aber als Zusammensteller hat man aber noch keine Möglichkeit dies zu tun.
Obwohl im Artikel 29 daruaf hingewiesen wird, dass auch die Behandlungseinheiten mit einer eigenständigen UDI-DI versehen werden müssen.
Wissen Sie vielleicht wann das Zeitfenster für die Registrierung für die Zusammensteller nach Artikel 22 geöffent wird.
Vielen Dank.
Mit freundlichen Grüßen
Davor Ninkovic
Lieber Herr Ninkovic,
bitte verzeihen Sie die ungewöhnlich lange Wartezeit.
Meine Kollegin Delia Yang hat mir die folgende Antwort gegeben:
Bezüglich des Zeitfensters lässt sich keine Angabe machen, sollte die Frage jedoch darauf abzielen, was zu tun ist, bis es diese Funktionalität in der EUDAMED gibt, gibt der BAnz AT 28.05.2021 B6 einen Hinweis in der i. Vorbemerkung (erste Zeile in der Tabelle)
https://www.bundesanzeiger.de/pub/publication/IGrRaNuJBWgwK7Giz8Y/content/IGrRaNuJBWgwK7Giz8Y/BAnz%20AT%2028.05.2021%20B6.pdf?inline
Herzliche Grüße
Anja Segschneider | Redaktion
Sehr geehrte Frau Segschneider,
Sehr geehrter Herr Klessascheck und Herr Seib,
vielen Dank für Ihre Antwort, Sie haben mir sehr geholfen.
Ihre Hilfe würde ich gerne erneut in Anspruch nehmen, da sich bei mir weitere Fragen aufgetan haben.
Für die Zusammenstellung des Kits packen wir einen Bestandteil des Kits um, d.h., dass das Produkt vom Karton in einen Polybeutel umverpackt wird. Der Originalzustand des Produktes an sich wird dabei nicht verändert- so mein Verständnis-. Anschließend werden alle Bestandteile des Kits in einen Karton zum Vertrieb verpackt.
Ist dieses Umverpacken zu verstehen im Sinne von Art. 16,Abs.2 (b) ? Bleiben wir somit der Zusammensteller und rutschen nicht in die Rolle des Herstellers?
Nochmal vielen Dank im Voraus für Ihre Hilfe!
Liebe Frau Graß,
ich antworte Ihnen per Email. Liebe Grüße, Mario Klessascheck
Sehr geehrte Frau Segschneider,
ich beziehe mich auf ihr Kommentar vom 04.02.2022.
Ich bin gerade auf eine widersprüchliche Information in der Art 22 MDR gestoßen.
Nach Art. 22 (1) können neben Medizinprodukten auch
„c) sonstige Produkte, die den für sie geltenden Rechtsvorschriften entsprechen, nur dann, wenn sie im Rahmen eines
medizinischen Verfahrens verwendet werden oder wenn ihr Vorhandensein im System oder in der Behandlungseinheit anderweitig gerechtfertigt ist.“
in eine Behandlungseinheit kombiniert werden.
Dabei steht unter Punkt c) explizit nicht nochmal sonstige Produkte „mit CE-Kennzeichnung“ wie unter Punkt a) und b), obwohl es dort offensichtlich ist. Damit sind ebenfalls, wie Sie oben beschrieben haben, Nicht-Medizinprodukte, die einer EU-Verordnung/Richtlinie folgen, verwendbar (z.B. Cosmetics EC 1223/2009), sofern ein medizinischer Nutzen für das System oder die Behandlungseinheit gerechtfertigt ist. Diese tragen kein CE-Kennzeichnen.
Nach Art. 22 (4) müssen aber alle Komponenten in der Behandlungseinheit eine CE-Kennzeichnung tragen, ansonsten wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen.
-> Schließt Art 22 (1)c nicht die Einschränkung unter Art. 22 (4) nicht aus oder bezieht sich in Art. 22 (4) „Produkte, die keine CE-Kennzeichnung tragen“ nur auf Produkte Medizinprodukte (MDR, IVDR etc.).
____
Des Weiteren frage ich mich, ist die erstmalige Abgabe des Systems- oder der Behandlungseinheit nicht auch ein „Inverkehrbringen“ („„[…]die erstmalige Bereitstellung eines Produkts,[…] auf dem Unionsmarkt“) der ganzen System- oder die Behandlungseinheit, denn das Art. 22 Produkt kann als eigenständiges Produkt angesehen werden (UDI-DI)? Die Art. 22 Produkte sind ja nach der Definition (MDR) „eine Kombination von zusammen verpackten und in Verkehr gebrachten Produkten, die zur Verwendung für einen spezifischen medizinischen Zweck bestimmt sind“?
D.h. die Behandlungseinheit nutzt bereits in Verkehr gebrachte Produkte und stellt diese anschließend nur noch „bereit“. Aber die Behandlungseinheit an sich, wird nochmal In Verkehr gebracht unter einem neuen Namen.
Vielen Dank für die Erklärung
Mit freundlichen Grüßen,
Marco Salis
Lieber Herr Salis,
Sie haben recht, das ist ein wenig verwirrend. Der Begriff „sonstige Produkte“ ist in 1a) und 1c) unterschiedlich zu verstehen. Bei 1a) ist ein Produkt gemäß Artikel 1, Absatz 4 ein Medizinprodukt, ein Zubehör oder ein Produkt gemäß Anhang XVI, das den Anforderungen der MDR genügen muss. In 1c) ist mit „Produkt“ ein Produkt im umgangssprachlichen Sinn gemeint, das nicht den Anforderungen der MDR unterliegt, aber konform mit anderen regulatorischen Anforderungen sein muss und daher ggf. auch eine CE-Kennzeichnung trägt. Absatz 4 erwähnt erneut den Begriff „Produkte“. Damit sind aber die Produkte in der Definition des Artikels 1, Absatz 4 gemeint. Das könnten z.B. FDA-zugelassene Medizinprodukte sein.
Zudem spricht der Artikel 22 von einer Inverkherbringung, da für diese Kombination eine neue Sache entsteht, die in dieser Kombination erstmals bereitgestellt wird.
Herzliche Grüsse, Mario Klessascheck
Guten Tag,
– Welche Übergangsfristen gelten für das Anbringen von UDI bei Systemen/Behandlungseinheiten?
– Gibt es neben dem Anbringen von UDI und Namen, Adresse, usw. gemäß Artikel 22, Absatz (5) noch weitere Anforderungen an die Kennzeichnungen von Systemen/Behandlungseinheiten?
Vielen Dank im Voraus für Ihre Rückmeldung!
Mit freundlichen Grüßen,
Caroline Anastasopoulos
Liebe Frau Anastasopoulos,
Es gibt keine Übergangsfrist für die Aufbringung der UDI-Träger bei Systemen oder Behandlungseinheiten, zumindest ist keine Frist definiert. Auch die MDCG Guidances 2018-3, 2018-4 und 2022-07 gehen darauf nicht ein. Weitere Anforderungen gibt es ebenfalls keine. Beachten Sie bitte dennoch die genannten MDCG Dokumente.
Liebe Grüsse, Mario Klessascheck
Schönen guten Tag,
ich hätte eine Frage bezüglich Artikel 22 „Einzelstücken“.
Was kann der Betreiber unternehmen, um selber einfach ein „Einzelstück“ zu betreiben?
D.h. der z.B. Philips Anlage geht kaputt und ein Händler bringt einen neuen Generator und eine Röhre von einem anderen Hersteller und der „Betreiber“ will dies auf eigene Verantwortung so betreiben.
Der Betreiber kann dies dann als „Einzelanfertigung“ oder mit Bauartzulassung agieren.
Handelt es sich hierbei überhaupt um ein Artikel 22 System?
Mit freundlichen Grüßen
Liebe Frau Straub,
Wenn eine Gesundheitseinrichtung mehrere Medizinprodukte (mit CE-Zeichen) zu einem neuen Produkt kombiniert und wenn sie dabei diese Produkte in ihrer ursprünglichen Zweckbestimmung verwendet (und damit diese Kombination im Sinne der jeweiligen Hersteller ist), hat die Gesundheitseinrichtung kein neues Produkt kreiert, es hat keine Eigenherstellung vorgenommen.
Wenn hingegen Produkte kombiniert werden, ohne dass diese Kombination vom Hersteller des Medizinprodukts vorgesehen wurde, ist eine Eigenherstellung zu vermuten. Artikel 22 greift insofern nicht, da keine Inverkehrbringung stattfindet. In diesem Artikel haben wir die verschiedenen Bedingungen schon einmal durchleuchtet. Vielleicht hilft das schon bei der Beantwortung Ihrer Frage. Falls nicht, fassen Sie gern nochmals nach.
Liebe Grüsse Mario Klessascheck
Ich habe eine allgemeine Verständnisfrage:
Ich habe den Eindruck, dass es mittlerweile viele Behandlungseinheiten auf dem Markt gibt, bei denen der Verantwortliche (für das Zusammensetzen von Systemen oder Behandlungseinheiten) kein CE-Zertifikat hat. Behandlungseinheiten werden nicht CE-gekennzeichnet, wenn sämtliche Bestandteile ein CE tragen und somit rechtmäßig in Verkehr gebracht wurden. Kann der Verantwortliche Behandlungseinheiten-Zusammensetzer demnach Produkte jeder Risikoklasse in einer Behandlungseinheit zusammenstellen, ohne ein entsprechendes CE-Zertifikat einer benannten Stelle zu haben? Weil im Grunde die Produkte von Herstellern mit entsprechenden CE-Zertifikaten bezogen werden? Oder kann zum Beispiel ein Hersteller von Klasse Is Produkten darüber hinaus Behandlungseinheiten herstellen, deren Bestandteile Medizinprodukte der Klasse IIa und IIb enthalten, ohne eine Anpassung seines CE-Zertifikats von Is auf IIb vornehmen zu müssen?
Mit freundlichen Grüßen
Lieber Herr Kienel,
Sofern die Produkte in einer für diese Produkte vorgesehenen und vereinbaren Weise zusammensetzt werden, können damit Systeme und Behandlungseinheiten erstellt werden. Der/die Zusammensteller:in muss allerdings eine entsprechende Erklärung gemäss Artike 22 abgeben. Eine Anpassung der Klassifikation wäre nur dann notwendig, wenn sich aus der Kombination neue Risiken ergeben, weil die Zusammenstellung so nicht vorgesehen war. Dann ist diese Kombination wie ein neues Produkt zu betrachten und muss neu klassifiziert werden.
Herzliche Grüsse, Mario Klessascheck
Sehr geehrtes Team des Johner Institut
Wir bieten ein System für die Anwendung in der Orthopädie an. In der CH sind wir lediglich Importeur und Händler und lassen die einzelnen Instrumente aus dem System von einem Hersteller in Deutschland fertigen. Die Einzelkomponenten haben alle ein CE-Zeichen, sind aber in der kombinierten Anwendung vorgesehen und somit als System zugelassen.
Wir erhalten die Einzelkomponenten vom Hersteller einzeln verpackt. Nun stellt sich die Frage, ob wir durch das reine Auspacken und zusammenstellen des Sets (i.e. Einlegen in die vorgesehenen Halterungen des Sterilisations-Siebs) in die Rolle des Herstellers rutschen. Handelt es sich hier um eine Umverpackung nach Art. 16 Abs. 2 Buchstabe b der EU-MDR?
Vielen Dank & Grüsse
S. Kramer
Liebe Frau/Herr Kramer,
Mein Kollege Christopher Seib gab mir die folgende Antwort: Wenn Sie die Produkte im Rahmen der Zweckbestimmung umpacken oder zusammenstellen, werden Sie nicht zum Hersteller. Wenn Sie dafür aber die Primärverpackung beschädigen/öffnen oder verändern müssen, dann sollten Sie die schriftliche Zustimmung des Herstellers einholen und prüfen, ob die Handlung möglicherweise einen Einfluss auf die Validierungsdaten wie Haltbarkeit oder Stabilität haben. Falls das der Fall wäre, übernehmen Sie Verantwortung als Hersteller.
Liebe Grüsse, Mario Klessascheck
Hallo Johner-Team.
wir haben ein System, welches aus zwei Geräten besteht (Gerät A und B) . Das eine Gerät (Gerät A) kann mit verschiedenen Modulen (1,2 oder 3) ausgestattet werden. Diese Module werden noch im Herstellungsprozess eingebaut und sind somit nicht durch einen Anwender austauschbar.
Ist es möglich, das System z.B. als „System 1“ aus „Gerät A“, „Gerät B“ und „Modul 2“ zu betrachten?
Oder müsste man das „Gerät 1“ aufteilen in: „Gerät A mit Modul 1“ und „Gerät B mit Modul 2“ und somit das oben genannte Beispiel „System 1“ als System aus „Gerät A mit Modul 1“ und „Gerät B“ betrachten?
Herzlichen Dank!
Lieber Leser,
Ich bin nicht sicher, ob ich die Kombinationen richtig erfasst habe. Wenn Sie unterschiedliche Varianten eines Moduls in ein Gerät/Endprodukt einbauen, ergibt sich daraus kein System im Sinne vom Artikel 22. Das gebrauchsfertige Gerät mit dem verbauten Modul erhält dann ein einzelnes CE. Sie müssen die Module daher nicht in die Systemvarianten einbeziehen.
Liebe Grüsse, Mario Klessascheck
Lieber Herr Klessaschek,
vielen Dank für die schnelle Antwort.
Es sind unterschiedliche Module, welche man in ein Gerät einbauen kann. Dieses Gerät ergibt zusammen mit einem weiteren Gerät das System nach Artikel 22.
Da es aber mehrere Module gibt, kam die Frage auf, ob man hierfür für jede Gerätevariante (Gerät + Modul) ein CE Kennzeichen braucht oder ob es möglich wäre, auch das Modul mit einem CE zu versehen und ob dies dann gemeinsam mit dem Gerät (in dem es verbaut wird) und dem zweiten Gerät ein System nach Artikel 22 bilden kann.
Wären die Module separat an das Gerät anbringbar, wäre das vermutlich in Ordnung. Aber in unserem Fall ist das Modul in einem der Geräte verbaut. Also das System würde schon vom Hersteller so verbaut werden.
Ich hoffe es wird nun klarer.
Viele Grüße
Liebes Johner-Team.
wir haben als Medizinproduktehersteller ein Legesystem (Klasse Is) und einen dazugehörigen Stent (Klasse IIb) als Einzelprodukte unter MDD zugelassen. Nun würden wir gerne das Legesystem mit diesem Stent vorladen und diese Kombination als Behandlungseinheit unter Artikel 22 Erklärung vertreiben. Ist dies zulässig?
Normalerweise werden das Legesystem und der Stent nach ihrer Produktion einzeln verpackt, sterilisiert und als 2 einzelne Produkte vertrieben. Für den Vertrieb als Behandlungseinheit würden wir im Rahmen der Produktion beide Medizinprodukte zusammensetzen (den Stent manuell auf den Führungskatheter des Legesystems führen), in eine gemeinsame (Primär-) Verpackung überführen und gemeinsam sterilisieren. Die Steri-, Verpackungs- und Transportvalidierung für das Set ist vorgesehen oder wird ggf. über Worst-Case Produkte abgedeckt.
Ist dieses Vorgehen noch konform zum Artikel 22 oder führt diese Prozessänderung dazu, dass es sich nun um ein neues Produkt handelt?
Recht herzlichen Dank im Voraus und beste Grüße
Valerie Herrmann
Liebe Frau Herrmann,
Ich habe mir für die Antwort Hilfe bei meinem Kollegen Luca Salvatore (Experte für Artikel 22) geholt. Er sagt:
Da Sie die beiden Produkte in einer mit der Zweckbestimmung vereinbaren Weise kombinieren, können Sie den Artikel 22 zusammen mit 2a-c) anwenden. Die Schwierigkeit liegt im Absatz (4). Wenn Sie die Sterilisation nicht gemäß den Anweisungen des Herstellers durchführen können, könnten Sie die Validieren den einzelnen Produktakten zuordnen, so dass kein neues Produkt geschaffen werden muss und Artikel 22 Anwendung finden kann.
Liebe Grüsse, Mario Klessascheck
Hallo,
wir haben einen ganz ähnlichen Fall. Wir sind Hersteller von Einzelprodukten, welche wir aber auch in eine Box packen und gemeinsam als Behandlungseinheit verkaufen (nicht in der Verpackung des Einzelproduktes). Uns stellt sich jedoch die Frage der Kennzeichnung. Gemäß Anhang VI Teil C 6.3.1. müssen alle Produkte in einer Behandlungseinheit auch eine UDI-Kennzeichnung tragen.
Wie würden Sie die den oben genannten Stent mit dem Führungskatheter kennzeichnen, wenn diese schon zusammengesetzt sind (UDI-DI/Artikelnummer/LOT/Haltbarkeit etc.)?
Liebe Frau Knieps,
das ist eine spannende und auch knifflige Frage, die ich intern mit einem Kollegen besprochen habe.
Wenn die einzelnen Produkte nicht über die erforderlichen Informationen verfügen, die gemäß Artikel 22(2) b) beigefügt werden müssen und normalerweise auf den Primärverpackungen der Einzelprodukte zu finden sind, wäre ein Inverkehrbringen gemäß Artikel 22 nicht möglich. Dadurch würde ein neues eigenständiges Produkt entstehen. Falls Sie der Hersteller aller Einzelkomponenten sind, besteht auch keine Notwendigkeit, die Produkte als eine Behandlungseinheit zu vertreiben. Durch das Zusammenstellen der Produkte im Rahmen ihrer eigenen Zweckbestimmung dürfen Sie die Produkte als Sachgesamtheit versenden. Sie sollten für die Kombination eine kurze technische Dokumentation erstellen und darin auf die Einzelakten verweisen.
Hilft Ihnen das weiter? Sonst fassen Sie gern nochmals nach. Schreiben Sie mich auch gern direkt an.
Liebe Grüsse, Mario Klessascheck
Liebes Johner-Team,
wir sind ein Hersteller für u.a. Legesysteme (Klasse Is) und Stents (Klasse IIb), die sowohl einzeln als auch zusammengesetzt als Behandlungseinheiten gemäß Artikel 22 vertrieben werden. Sowohl die Legesysteme als auch die Stents haben eine eigenständige Produktakte.
In Hinblick auf die Behandlungseinheit stellt sich die Frage, wie es sich mit der klinischen Bewertung und dem PMS verhält.
– Wird die Behandlungseinheit im PMS/klinischen Bewertung der Einzelprodukte betrachtet? Oder
– Soll die Behandlungseinheit eigenständig behandelt werden?
Vielen lieben Dank im Voraus und beste Grüße
Sabrina
Liebe Frau Weinkauf,
Für eine Behandlungseinheit nach Artikel 22 müssen Sie keine zusätzliche klinische Bewertung durchführen. Die Produkte wurden ja bereits mit der vereinbaren Zweckbestimmung geprüft. Sie müssten somit die Punkte 2 a) bis c) betrachten. Die Frage bezüglich PMS ist interessant. Da Sie der Hersteller für beide Produkte sind, sollten Sie auf Ebene der Einzelprodukte ebenfalls deren bestimmungsgemässe Verwendung mit anderen Produkten überwachen. Somit wäre aus meiner Sicht ein separater PMS Plan nicht notwendig.
Hilft Ihnen das weiter?
Liebe Grüsse, Mario Klessascheck
Liebes Johner-Team,
Benötigen Behandlungseinheiten bestehend aus MDD-Produkten weiterhin eine Erklärung nach §12 MDD oder bereits nach Art. 22 MDR?
Herzlichen Dank!
Liebe Frau Mirschberger,
Sie benötigen eine Erklärung nach Artikel 22. Dabei spielt es keine Rolle, ob eins der Produkte noch unter MDD in Verkehr gebracht wurde.
Liebe Grüsse, Mario Klessascheck
Lieber Herr Klessascheck,
hierzu hätte ich eine Rückfrage. Im MDCG 2021-25 (Tabelle, Seite 9) wird bezüglich Art. 22 darauf verwiesen, dass dieser nur zutrifft, wenn legacy devices und mind. 1 MDR device kombiniert werden. Dies würde bedeuten, dass alleinige MDD Produkte nur unter Art 12 Erklärungen laufen dürfen. Wie bewerten Sie diese Information der MDCG?
Wir haben derzeit ausschließlich MDD Produkte, die wir gemäß MDD Art 12 als Behandlungseinheit kombinieren wollen. Dürfen wir überhaupt noch Art. 12 Erklärungen ausstellen?
Herzlichen Dank und viele Grüße
Liebe Frau Marquard-Handreck,
Vielen Dank für Ihre Geduld. Mein Kollege Christopher Seib, unser MDR Experte, war in den Ferien. Er hat mir wie folgt geantwortet.
Der Sachverhalt hat nur indirekt etwas mit den Systemen zu tun. Man darf nur Systeme unter der MDR in Verkehr bringen, wenn zumindest ein Medizinprodukt darin die MDR erfüllt. Die MDD ist nicht länger gültig und Hersteller dürfen keine neuen Produkte unter der MDD in Verkehr bringen. Ein System nach Artikel 12 wäre aber ein neues Produkt, das gemäß 12(2) zunächst noch nicht als in Verkehr gebracht zählt, auch wenn es die einzelnen Produkte darin eigentlich schon sind. Deswegen gibt es für MDD Artikel 12 Erklärungen keine Möglichkeit, diese noch auszustellen.
Hilft Ihnen die Antwort weiter? Sonst fassen Sie gern noch einmal nach.
Liebe Grüsse
Mario Klessascheck
Hallo liebes Johner-Team,
vielen Dank für den interessanten Artikel.
Ich beschäftigte mich seit längerem mit dem Thema Self-Collection-Kits/ Home Tests für verschiedene BioMarker. In diesem Bereich gibt es bereits zahlreiche Anbieter auf dem Markt, von denen ich schon einige getestet habe.
Nachdem ich Ihren Artikel gelesen habe, würde mich interessieren, wie es die Anbieter schaffen „System- und Behandlungseinheiten“ mit einer Kombination von Medizinprodukten auf den Markt zu bringen, welche laut Zweckbestimmung nur für die Anwendung durch medizinisches bzw. Fachpersonal ausgelegt sind.
Ich würde mich über einen Austausch freuen.
Beste Grüße
M
Liebe:r M
ich bin mir leider nicht ganz sicher, ob ich Sie richtig verstanden habe und meine folgende Ausführung Ihre Frage beantwortet. Sollte dies nicht der Fall sein, melden Sie sich gerne nochmals bei uns, zum Beispiel über das kostenlose Micro-Consulting.
Sollten sich in der Behandlungseinheit Produkte befinden, die laut Zweckbestimmung nur durch medizinisches Fachpersonal anzuwenden sind, ist auch die Behandlungseinheit nur durch entsprechendes Fachpersonal zu verwenden. Eine Anpassung der Zweckbestimmung ist nicht möglich. Dies geht insbesondere aus MDR Artikel 22 Absatz 1 Satz 1 und Absatz 2 a) und b) hervor.
Bei Anpassung der Zweckbestimmung der zusammen verpackten Produkte, kann das Set nicht mehr als Behandlungseinheit in Verkehr gebracht werden, und muss stattdessen als Medizinprodukt oder IVD (Abhängig von Inhalt und neuer Zweckbestimmung) einer Konformitätsbewertung unterzogen werden.
Herzliche Grüße,
Ulrich Hafen
Liebes Johner Team,
Wir möchten ein Set im Sinne von MDR Artikel 22 (1) auf den Markt bringen. Ein Produkt im Set ist steril (Primärverpackung ist ein Kunststoffbeutel; Sekundärverpackung ist ein Pappkarton). Wir beziehen dieses Produkt als Handelsware. Im Rahmen des Set Packings würden wir dieses Produkt unverändert ihrer Original Box entnehmen und in ihrer Original Primärverpackung in die Sekundärverpackung des Sets überführen. Die Aufrechterhaltung der Sterilbarriere durch das Umverpacken würden wir durch entsprechende Maßnahmen sicherstellen. Transportvalidierungen für das Set sind natürlich auch vorgesehen.
Das Umpacken von Handelsware durch einen Händler ist gemäß Artikel 16 (2) b) zulässig. Greift das auch für den Setpacker? Zudem wäre die Frage ob dann auch Artikel 16 (4) greift, das ja besagt, dass die Umverpackung einer Meldepflicht an Hersteller und Behörde unterliegt. Gilt dies auch für Sets gemäß Artikel 22 (1)?
Ganz herzlichen Dank im Voraus für Euren Support!
Liebe Frau Herrmann,
Ihre Frage habe ich intern weitergeleitet und mein geschätzter Kollege Christopher Seib hat mir folgendermassen geantwortet: Artikel 16 gilt gemäß Überschrift auch für „andere Personen“ – damit sind explizit auch die „Setpacker“ gemeint, die in Artikel 22 als andere Personen bezeichnet werden. Das Öffnen der Sekundärverpackung eines sterilen Produktes stellt erstmal eine konformitätsrelevante Änderung dar. Da die Primärverpackung nicht verändert wird, greifen Artikel 16 Absatz 3 und 4 für die besonderen Händlerpflichten.
Vom BVMed (Wichtig für MedTech-Händler | Neuer BVMed-Leitfaden zur Vereinzelung von Medizinprodukten nach der MDR – BVMed) gibt es dazu einen sehr schönen Leitfaden.
Hilft Ihnen das weiter?
Herzlichst, Mario Klessascheck
Liebes Johner Team,
danke für den sehr guten Fachartikel und die überaus hilfreichen Antworten auf die Fragen aus der Praxis.
Folgende Frage: laut MDR Artikel 2 (10) muss ein Procedure Pack für einen bestimmten medizinischen Zweck bestimmt sein. Wie verhält es sich, wenn eines der Produkte des PP ein Annex XVI Produkt ohne medizinische Zweckbestimmung ist und auch die Zweckbestimmung des gesamten PP keine medizinische ist? Gibt es in diesem Fall überhaupt die Möglichkeit, das Annex XIV Produkt gemeinsam mit einem CE konformen MD als ein PP gemeinsam in Verkehr zu bringen bzw. was wäre die Alternative für den Fall, dass dies nicht möglich ist?
Herzlichen Dank.
T
Laut Definition muss das PP einem gemeinsamen medizinischen Zweck dienen. Wenn dies nicht der Fall ist, d.h. die Kombination keine medizinische Zweckbestimmung hat, liegt kein PP im Sinne der MDR und Artikel 22 vor.
Somit wäre Artikel 22 nicht anwendbar. Für eine genauere Betrachung würden wir detaillierte Informationen zum Zweck der Kombination sowie der einzelnen Produkte benötigen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
danke für die rasche und kompetente Rückmeldung; da die Legaldefinition eines PP nach MDR in dem beschriebenen Fall aufgrund der nicht-medizinischen Zweckbestimmung nicht erfüllt ist – als was würden Sie denn die Kombination eines Annex XVI Produktes mit einem Medizinprodukt bezeichnen bzw. welche regulatorischen Vorgaben wären für dieses Produkt einzuhalten? Im konkreten Fall kann das Annex XVI Produkt ausschließlich unter Verwendung eines (beigepackten), CE- gekennzeichneten Medizinproduktes verwendet werden.
Beste Grüße
T
Guten Tag,
diese Frage ist schwer, pauschal zu beantworten. Denn es ist ja nicht ausgeschlossen, dass eine Kombination aus Medizinprodukt und Anhang XVI-Produkt nicht doch einem spezifischen medizinischem Zweck dienen kann. Wenn dies nicht gegeben ist, wie in Ihrem Fall, sehe ich an die „Sachgesamtkeit“ keine zusätzlichen spezifischen Verpflichtungen gemäß MDR. Jedes Produkt muss die anwendbaren Anforderungen der MDR erfüllen. Sicherlich ist bei dem Zusammenspiel verschiedener Produkte die Anforderung 14.5 relevant (Produkte, die gemeinsam mit anderen Produkten oder Produkten, die keine Medizinprodukte sind, eingesetzt werden sollen, werden so ausgelegt und hergestellt, dass das Zusammenspiel und die Kompatibilität zuverlässig und sicher sind.).
Freundliche Grüße
Luca Salvatore
In einer Diskussionsrunde kam die Frage auf, nach welcher DIN Norm sich eine Zusammensetzer von Behandlungseinheiten Artikel 22 zertifizieren muss. Die EU-VO 2017/745 gibt da keine konkrete Hinweise.
Einige in der Runde meinten man sollte nach der DIN EN ISO 13485 zertifiziert sein, andere wiederum sagte die DIN EN ISO 9001würe langen.
Lieber Herr Ninkovic,
vielen Dank für die Frage. Mein Kollege Christopher Seib (unser Experte für Artikel 22) hat mir folgendes dazu gesagt. Systeme nach Artikel 22 sind keine Medizinprodukte. Die Artikel 22 Person ist entsprechend auch kein Hersteller und benötigt dafür auch nicht zwingend ein QMS. Wenn die Produkte noch sterilisiert werden, oder konformitätsrelevante Änderungen durchgeführt werden, wird die Person zum Hersteller. Hersteller müssen ein QMS haben, ohne Vorgabe eines Standards. QMS System müssen ab Klasse IIa Produkte zertifiziert werden.
Herzliche Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
vielen Dank für die rasche Rückmeldung.
Verstehe ich das dann richtig, dass ein Zusammensetzer nach Artikel 22 zum jetzigen Zeitpunkt keinerlei QMS benötigt, weder nach 9001 noch 13485, sofern er keine Produkte sterilisiert oder konformitätsrelevante Änderungen vornimmt.
Wenn konformitätsrelevante Änderungen nach Artikel 16 (2) (b) durchgeführt würden, welches QMS müsste man nach Artikel 16 (3) als Nachweis erbringen? Langt ein QMS nach 9001 oder benötigt man doch ein QMS nach 13485?
Herzliche Grüße
Die beiden Artikel 22 hängt nur indirekt mit Artikel 16 zusammen. Wenn Sie Händler sind, unabhängig davon, ob Sie Systeme bauen, müssen Sie ein QM System betreiben. Das QM System kann nach ISO 13485 oder ISO 9001 oder ohne Norm aufgebaut sein, da Sie es nicht zertifizieren müssen. Es macht aber sicherlich Sinn für die verlangten Prozesse, die beiden Normen als Vorlage zu nehmen, die sich in vielen Bereichen auch sehr ähnlich sind. Mit einer 9001 liegen Sie sicher nicht falsch.
Liebe Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
benötigt der Assembler ein QMS (Abs. 3 und 4 Artikel 22 MDR trifft nicht zu)? und muss das Hersteller-Symbol auf die Verpackung, obwohl der Assembler im Sinne der MDR kein hersteller ist?
Vielen Dank!
Sehr geehrte Frau Rau,
danke für Ihre spannende Frage! Ich gehe bei meiner Antwort davon aus, dass Sie dies unter Assembler verstehen.
Aus regulatorischer Sicht ist ein Assembler eine Software im Rahmen eines QM-relevanten Prozesses. Damit kann das Kapitel 4.1.6 der ISO 13485 gelten. Der Artikel 22 ist nicht anwendbar, weil der Assembler kein Produkt oder Teil eines Produkts ist.
Ob Sie den Assembler validieren müssen, hängt von den Umständen / Risikomanagement ab. In der Regel ist das nicht notwendig, weil das Endergebnis der Assemblings geprüft wird.
Beste Grüße, Christian Johner
Guten Tag,
hoffe ihr könnt bei meiner Frage weiterhelfen.
Wir stellen Procedure Packs her, beide Komponenten sind noch nach MDD zertifiziert, eine Declaration of Content gemäss Artikel 12 MDD ist vorhanden.
Da wir unsere PPs in unterschiedlichen Märkten verkaufen, heisst das selbe Zubehör unterschiedlich.
Nun haben wir das Problem, dass wir von einer Komponente nicht mehr genügend auf Lager haben, um den PP zu bestücken. Wir hoffen, das spezifikationsgleiche Zubehör aus dem anderssprachigen PP verwenden zu können, wissen aber nicht, wie wir hier gesetzeskonform vorgehen sollen.
Eine neue Bescheinigung nach Artikel 22 MDR ist nicht möglich, da keine der Komponenten nach MDR zertifiziert ist. Eine neue Bescheinigung nach Artikel 12 MDD ist nicht möglich, da eine solche nicht mehr ausgestellt werden darf.
Könnten wir uns hier eventuell mit einer „nicht wesentlichen Änderung“ behelfen?
Ich danke schon im voraus für eure Zeit und Hilfe
mfG
Eva
Liebe Frau Albertini,
vielen Dank für die spannende Frage, deren Beantwortung ist nicht ganz einfach ist. Ich habe mich dazu mit meinem Kollegen Christopher Seib ausgetauscht und seine Meinung dazu lautet wie folgt.
Eine Neuzulassung ist nicht möglich, da gemäss altrechtliche Produkte (dazu zählen auch Procedure Packs) nur dann noch weiter in Verkehr gebracht werden können, wenn sie weiterhin die Anforderungen der Richtlinie erfüllen und nicht signifikant geändert wurden. Das MDCG Guidance Dokument 2020-3 beschreibt den Umgang mit Änderungen. Wenn in Ihrem Fall nur eine Namensänderung am Produkt erfolgt und das restliche Labeling (Symbole etc.) identisch bleibt, das entspricht einem Design Change nach Chart B, wäre die Änderung nicht signifikant. Die Rückverfolgbarkeit wäre nach wie vor gegeben und es entstehen keinerlei neue oder veränderte Risiken.
Hilft Ihnen das weiter? Wenn nicht haken Sie gern noch einmal nach.
Herzliche Grüsse, Mario Klessascheck
danke für die antwort herr klessascheck, und danke auch an herrn seib. ja, es wäre eine reine namensänderung – symbole, labelling, risiko, rückverfolgbarkeit… bliebe alles gleich.
das problem das ich mit dieser situation habe ist, dass in der declaration of content die produkte ja namentlich genannt werden. und wenn jetzt eines der produkte nicht mehr x sondern y heisst, steht in der delcaration aber weiterhin produkt x, da ich diese nicht mehr ändern darf.
wie würden sie mit dieser „herausforderung“ umgehen? einen annex hinzufügen?
nochmal danke für ihre hilfe!
Liebe Frau Albertini,
ich melde mich per Email bei Ihnen.
Liebe Grüsse, Mario Klessascheck
Sehr geehrte Kollegen,
ich habe eine klare aber schwierige Frage:
Problemstellung:
Ein Systemträger mit einer Mehrfachsteckdose wird mit insgesamt 6 Geräten bestückt. Wenn alle Geräte eingeschaltet werden, wird der Berührstrom von 500 µA nicht mehr eingehalten.
Lösungsidee:
Die 6 Geräte haben unterschiedliche Anwendungszwecke. Das bedeutet, sie werden so in Kombination nicht verwendet. Tatsächlich benötigt der Anwender entweder die oberen drei oder die unteren drei Geräte.
Die Frage:
Können wir die Definition des ME-Systems so auslegen, dass zwei ME-Systeme auf dem Systemträger verbaut sind? Dann könnte der Berührstrom für jedes ME-System bestehend aus Systemträger und drei Geräten einzeln gemessen werden (obwohl alle 6 Geräte auf dem Systemträger gestapelt sind). Im Verwendungshinweis wäre ein entsprechender Vermerk, dass nicht sämtliche Geräte gleichzeitig eingeschaltet werden dürfen.
Lieber Herr Pilz,
Der Berührungsstrom wird unter Normal Condition nur an Teilen gemessen, die nicht schutzleiterverbunden sind (siehe 8.7.4.6 a)). Daher sollte die Messung keinen Fehler ergeben. Wenn der Schutzleiter in der Mehrfachsteckdose unterbrochen wird, dann wird der Berührungsstrom an Teilen gemessen, die normalerweise mit dem Schutzleiter verbunden sind, da dann der Erdableitstrom zum Berührungsstrom wird. Wenn der Grenzwert überschritten wird, müssten Sie einen Trenntrafo oder einen weiteren Schutzleiter einplanen. Eine Aufteilung in zwei ME Systeme ist meines Wissens nicht zulässig oder gewünscht.
Falls ich Ihre Frage falsch verstanden habe, fragen Sie gern nochmal nach.
Liebe Grüsse, Mario Klessascheck
Guten Tag,
zunächst vielen Dank für den sehr informativen und klaren Artikel.
Ich habe eine Frage über die ich jetzt schon länger nachdenke. Müssen bei einer Behandlungseinheit die Hersteller der einzelnen Bestandteile erkenntlich sein, beispielsweise über das Label oder muss nur der Hersteller der Behandlungseinheit gekennzeichnet sein?
Liebe Frau Meier,
Vielen Dank für die interessante und wichtige Frage.
Der Artikel 22 verweist im Bereich der Kennzeichnung auf den Artikel 23 im Kapitel III. Dort steht im Absatz 23.1.: „Jedem Produkt werden die notwendigen Angaben beigefügt, die die Identifizierung des Produkts und des Herstellers ermöglichen, sowie alle für den Anwender oder gegebenenfalls dritte Personen relevanten Informationen über die Sicherheit und Leistung des Produkts. Diese Angaben können auf dem Produkt selbst, auf der Verpackung oder in der Gebrauchsanweisung angebracht sein und werden.“
Demnach müssen die Angaben nicht auf der Verpackung oder dem Produkt selbst erkenntlich sein. Wichtig ist aber, dass der Anwender die Komponenten identifizieren kann, um das Produkt gemäß Gebrauchsanleitung (können auch mehrere sein) sicher anwenden zu können.
Hilft Ihnen das weiter?
Liebe Grüsse Mario Klessascheck
Hallo,
ich habe eine Frage zu der Kombination von Produkten in einer Behandlungseinheit.
Der Artikel 22 schreibt: „[…] die PRODUKTE mit einer CE-Kennzeichnung mit folgenden anderen Medizin- oder sonstigen Produkten […] kombinieren, […] müssen eine Erklärung abgeben:
1. sonstige Produkte mit CE-Kennzeichnung;
2. […]
3. sonstige Produkte, die den für sie geltenden Rechtsvorschriften entsprechen, nur dann, wenn sie im Rahmen eines medizinischen Verfahrens verwendet werden oder wenn ihr Vorhandensein im System oder in der Behandlungseinheit anderweitig gerechtfertigt ist.“
-> Kann ich „PRODUKTE“ im ersten Satz so verstehen, dass MINDESTENS 2 Medizinprodukte nach MDR kombiniert werden müssen, da ier von Mehrzahl gesprochen wird, und ZUSÄTZLICH dann noch andere Medizinprodukte oder sonstige Produkte (Punkte 1-3) hinzugefügt werden können ODER reicht es aus, wenn nur eins der Medizinprodukte ein Produkt nach MDR ist (mit CE-Kennzeichnung) und die restlichen sonstigen Produkte zum Beispiel ohne CE-Kennzeichnung, aber dennoch den geltenden Rechtsvorschriften entsprechen (z.B. nach Punkt 3).
Beispiel:
Die Kombination eines MDR Klasse I Produktes mit einem Kosmetika (EU Verordnung 1223/2009). Ist diese Kombination zulässig, da hier nur 1x MDR und CE-gekennzeichnetes Produkt in der Behandlungseinheit enthalten ist.
Vielen Dank.
Mit freundlichen Grüßen,
Marco Salis
Lieber Herr Salis,
Vielen Dank für das Lesen unseres Blogs und Ihre spannende Frage. Sie haben es richtig verstanden, Artikel 22 lässt es zu Nicht-Medizinprodukte (sonstige Produkte) mit Mediznprodukten zu kombinieren, solange sie in einer der jeweiligen Zweckbestimmung entsprechenden Weise kombiniert werden. Eines der Produkte muss ein Medizinprodukt sein, anderenfalls wäre die MDR für die Kombination nicht zuständig. Hilft Ihnen das weiter? Sonst fragen Sie gern nach.
Liebe Grüsse, Mario Klessascheck
Hallo,
ich benötige auch Unterstützung im regulatorischen Dschungel von Behandlungseinheiten.
Wir kämpfen uns gerade durch den MDR Antrag und versuchen herauszufinden, wonach die benannte Stelle den Sampling Plan zur Prüfung der Technischen Dokumentation schreibt. Natürlich verweisen sie auf das MDCG Dokument 2019-13, welches meiner Meinung nach für Behandlungseinheiten nach Art. 22 der MDR nicht richtig passt. Natürlich ist dieser Posten im Angebot eine große Unbekannte. Können Sie mir helfen, wonach man die Behandlungseinheiten sampeln würde, wenn ja die Vergabe von einem MDN code nicht zulässig ist, da Behandlungseinheiten im Sinne der MDR keine Medizinprodukte sind?
Liebe Frau Kuhlmann,
Vielen Dank für Ihre Frage.
Ich bin noch nicht sicher, ob ich Ihre Frage richtig verstehe. Da Sie den Artikel 22 anbringen, vermute ich, dass Sie kein Produkt entwickeln, sondern zwei verkehrsfähige Produkte als System oder Behandlungseinheit vertreiben möchten. In dem Fall müssen Sie keine technische Dokumentation erstellen und Sie benötigen auch keine benannte Stelle für die Überprüfung. Wenn Sie mit Ihre Situation genauer schildern, kann ich konkreter antworten.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck, herzlichen Dank für die Rückmeldung.
Richtig, wir kaufen verkehrsfähige Medizinprodukte, packen diese nach Kundenwunsch in Behandlungseinheiten, sterilisieren diese mittels ETO (daher wird die benannte Stelle benötigt) und bringen diese als sterile Behandlungseinheiten nach Art. 22 inverkehr.
Nun haben wir den MDR Antrag gestellt und wollen abschätzen, was von denen in Bezug auch die Technische Dokumentation geprüft wird. Aktuell haben wir ein EG-Zertifikat gem. 93/42 EWG Anhang V.
Liebe Grüße
Jessica
Liebe Frau Kuhlmann,
Dass Sie sterilisieren, hatte ich vermutet. In dem Fall ist die Beteiligung der Benannten Stelle auf die Aspekte des Sterilisationsverfahrens beschränkt. Das betrifft im Wesentliche die folgenden Aspekte:
Von meinem Kollegen Christopher Seib habe ich noch die folgenden Punkte erhalten
Mein Rat wäre noch, dass Sie einfach die benannte Stelle fragen. Die können Ihnen genau sagen, was sie sehen möchten und haben manchmal fertige Checklisten.
Hilft Ihnen das weiter?
Mit vorweihnachtlichen Grüßen,
Mario Klessascheck
Guten Tag Herr Klessascheck,
wir sind Inverkehrbringer steriler Behandlungseinheiten und verfolgen mit großem Interesse Ihren Blog. Vielen Dank auch für diesen informativen Beitrag für uns Setpacker. Aktuell befinden wir uns in der Umstellung auf MDR Artikel 22. In der Dokumentenerstellung orientieren wir uns maßgeblich an den Question & Answers von MedTech Europe, Ausgabe 2.0 aus März 2020. Insbesondere in Bezug auf den Inhalt des Procedure Pack Files halten wir uns an die aufgelisteten Dokumente. Eine besondere Fragestellung hat sich hinsichtlich des anzuwendenden Konformitätsbewertungsverfahrens ergeben, das, gem. Artikel 22 (3), sich lediglich auf die Aspekte des Sterilisationsverfahrens beschränkt, die der Gewährleistung der Sterilität des Produkts bis zur Öffnung oder Beschädigung der Verpackung dienen. Ist diese Einschränkung so zu verstehen, dass alle Dokumente rund um die Themen Verpackung, Verpackungsvalidierung, Sterilisation, Sterilsationsvalidierung, Shelf life, Shelf life Validierung im Fokus dieses Konformitätsbewertungsverfahren stehen? Sicherlich werden auch allgemeine Punkte wie die Erklärung nach Artikel 22 sowie die Kennzeichnung Gegenstand der Begutachtung durch die Benannte Stelle sein. Wie sieht es mit evtl. weiteren allgemeinen Dokumenten aus, die in einem vollständigen Konformitätsbewertungsverfahren eingereicht werden müssten, wie z.B. Risikomanagement oder die Grundlegenden Sicherheits- und Leistungsanforderungen? Sind diese auch in unserem Fall Teil des Konformitätsbewertungsverfahrens, aber nur bezogen auf die o.g. Kriterien oder sind diese Dokumente nicht Gegenstand des Konformitätsbewertungsverfahrens? Wie sieht es aus mit der klinischen Bewertung oder PMS? Können diese für uns entfallen?
Vielen Dank im Voraus für Ihre Hilfe.
Mit freundlichen Grüßen
Sinah Wendt
Hallo Frau Wendt, genau diesen Fragen müssen wir uns auch gerade stellen und leider bekommen wir auch kaum Klarheit in dem MDR Dschungel.
Sie erwähnen das Question & Answers von MedTech Europe, Ausgabe 2.0 aus März 2020 als Quelle. Diese würde mich mir gerne einmal anschauen, kann aber im Netz hierzu leider nichts finden. Könnten Sie mir verraten, wo ich mir dieses herunterladen kann?
Liebe Frau Wendt,
Vielen Dank, dass Sie den Blog lesen und vielen Dank für das nette Kompliment, dass mich sehr freut.
Die Punkte im Anhang IX beschränken sich auf die Aspekte des Sterilisationsverfahrens und die Gewährleistung der Sterilität. Dazu gehört auch, dass Sie in einer Prozessrisikoanalyse mögliche Verfahrensfehler von der Sterilisation bis zur Öffnung der Verpackung analysieren sollten. Sie sollten auch bewerten, ob ein möglicher Defekt vom Kunden erkannt werden kann. Eine klinische Bewertung müssen Sie nicht durchführen. Das PMS könnte auf die Punkte der Verpackung reduziert werden und sollte beinhalten, dass Sie die Meldungen der Hersteller der Produkte überwachen, zum Beispiel, wenn sich die Vorgaben für die Sterilisation ändern.
Liebe Grüsse und alles Gute für 2024, Mario Klessascheck
Lieber Herr Klessascheck,
ich verfolge natürlich auch sehr interessiert hier die Diskussion und möchte zu Ihrer letzten Rückmeldung gerne noch unsere persönliche Erfahrung aus dem letzte Audit durch die benannte Stelle teilen. Unsere Risikoanalyse sollte sich laut Aussage des TÜVs auf den gesamten Herstellprozess beziehen, sprich von der Erfassung der Kundenanforderungen bis zur Überwachung nach dem Inverkehrbringen.
Liebe Frau Kuhlmann,
vielen Dank dafür, dass Sie ihre Erfahrungen hier teilen. Das ist sehr hilfreich. Ein wenig überrascht bin ich von der Aussage des TÜVs, da Sie wohl kaum die Kundenanforderungen kennen können, die der Hersteller bei der Auslegung hatte. Es sei denn, Sie sind selbst Hersteller, dann verstehe ich die Anforderung.
Liebe Grüsse, Mario Klessascheck
Guten Morgen Herr Klessascheck,
in dem Fall geht es um die Kundenanforderungen an unsere Behandlungseinheiten. Welche Komponenten, welche Packreihenfolge, welche Einschlagmethode, steril oder unsteril etc. Dies soll in der Risikoanalyse bis hin zum PMS der Behandlungseinheiten abgedeckt sein.
Herzliche Grüße
Hallo,
Gemäß Artikel 22 Absatz 1(c) dürfen auch Produkte in Behandlungseinheiten verwendet werden, die nicht den Anforderungen der MDR unterliegen, aber konform mit anderen regulatorischen Anforderungen sein müssen und daher ggf. auch eine CE-Kennzeichnung tragen. Weiterhin heißt es in Absatz 4: Enthält das System oder die Behandlungseinheit Produkte, die keine CE-Kennzeichnung tragen,[…] so wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen.
Wann muss ich bei sonstigen Produkten denn kein eigenes Konformitätsbewertungsverfahren nach Artikel 52 durchführen? Wie sieht es Beispielsweise mit einer PE-Schürze (Wegwerfschürze) aus, die ein Anwender als persönliche Schutzausrüstung in einer Behandlungseinheit haben möchte, aber vom Hersteller nicht als zugelassenes Medizinprodukt verkauft? Ich würde gerne für diese Produktgruppe Anforderungen formulieren, die es dann gilt zu überprüfen, bevor man die Komponente zur Verwendung freigibt.
Liebe Frau Kuhlmann,
Vielen Dank, dass Sie den Blog lesen und vielen Dank für die interessante Frage.
Meine liebe Kollegin Katharina Keutgen hat mir folgende Antwort gegeben: Gemäß Artikel 22 (1) dürfen neben Medizinprodukten und In-vitro-Diagnostika mit CE-Kennzeichnung sonstige Produkte in Systemen und Behandlungseinheiten verwendet werden, wenn sie den für sie geltenden Rechtsvorschriften entsprechen und sie im Rahmen eines medizinischen Verfahrens verwendet werden oder ihr Vorhandensein anderweitig gerechtfertigt ist. Absatz (4) des Artikels 22 impliziert, dass eine CE-Kennzeichnung zum Nachweis mit den für die sonstigen Produkte geltenden Rechtsvorschriften vorliegen muss. Im Fall von persönlicher Schutzausrüstung müsste also eine CE-Kennzeichnung zum Nachweis der Konformität mit der Verordnung (EU) 2016/425 vorliegen. Ist dies nicht der Fall, muss das Produkt einem Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen werden, z.B. als Bestandteil des eigentlichen Medizinprodukts.
Herzliche Grüsse, Mario Klessascheck
Lieber Herr Klessascheck,
Wir bedanken uns ganz herzlich für Ihre Antwort auf unsere letzte Anfrage vom 03.01.2024. Uns beschäftigt zurzeit noch eine weitere Fragestellung. Als Hersteller für Behandlungseinheiten stellen wir individuelle OP-Sets nach Kundenwunsch zusammen.
Einige unserer Lieferanten bieten Einmalinstrumente unsteril zum Einbringen in Behandlungseinheiten und anschließenden Sterilisation an. Für uns stellt sich bei einigen Instrumenten die Schwierigkeit der Klassifizierung. Handelt es sich um unsterile chirurgische Einmalinstrumente, so sind sie in der Klasse IIa einzustufen und die Sachlage ist eindeutig.
Fragen ergeben sich bei den unsterilen Einmalinstrumenten, welche nicht invasive Instrumente sind, und die nicht zum Anschluss an ein Klasse II Produkt dienen. Hierbei handelt es sich unserer Ansicht nach um ein Vorprodukt/Zwischenprodukt. Der Lieferant kennzeichnet diese folgerichtig mit CE, aber ohne Angabe einer Benannten Stelle.
Unter der Medizinprodukterichtlinie MDD war ein Äquivalenzvergleich zur sterilen Version des Produkts möglich. Wie wird dieser Sachverhalt unter Artikel 22 MDR bewertet? Uns liegen die Sterilisationsfreigabe und die Sterilisationsvorgaben des Instrumentenherstellers vor.
Wie ist damit umzugehen, wenn es kein steriles Äquivalenzprodukt gibt? Auch in diesen Fällen liegen die Sterilisationsfreigabe und die Sterilisationsvorgaben des Instrumentenherstellers vor, allerdings ist zu bedenken, dass keine Benannte Stelle diese Anleitung geprüft hat, da es sich um unsterile Einmalinstrumente als Vorprodukt / Zwischenprodukt handelt. Wird dann aus Klasse I Vorprodukten ein Klasse Is Medizinprodukt? Was ist unter Artikel 22 MDR zu beachten, wenn wir diese Produkte in unsere Behandlungseinheiten einbringen möchten?
Vielen Dank im Voraus für Ihre Hilfe und freundliche Grüße
Sinah Wendt
Hallo Zusammen,
eine sehr interessante Frage, zu der ich ebenfalls eine Antwort suche. Zudem haben wir ein ähnliches Problem: Wir sind ebenfalls Hersteller von Behandlungseinheiten für den OP-Bereich. Wir kaufen ebenfalls fertige Medizinprodukte nach Kundenwunsch ein. Nun führen viele Hersteller zahlreiche Produkte nur noch als Industrieware, um Kosten zu sparen, stellen dann aber eine Übereinstimmungserklärung aus, dass diese Produkte unter gleichen Bedingungen (Anforderungen der 13485) und unter Verwendung der selben Materalien hergestellt werden. Sie werden aber nicht mit einem CE-Zeichen versehen. Somit würde für mein Verständnis ein eigenes Konformitätsbewertungsverfahren durchgeführt werden müssen oder wie sehen Sie das?
Liebe Frau Kuhlmann,
Artikel 22, Absatz 4 ist da sehr eindeutig. Ich zitiere: „so wird das System oder die Behandlungseinheit als eigenständiges Produkt behandelt und dem einschlägigen Konformitätsbewertungsverfahren gemäß Artikel 52 unterzogen. Die natürliche oder juristische Person ist den für die Hersteller geltenden Pflichten unterworfen.“ Das bedeutet, Sie müssten die Pflichten eines Herstellers übernehmen. Dazu müssten Sie Zugriff auf die technischen Unterlagen erhalten.
Hilft Ihnen das weiter?
Liebe Grüsse, Mario Klessascheck
Liebe Frau Wendt,
vielen Dank für Ihre spannende Frage. Ich habe das auch intern mit unserem Team diskutiert. Wenn der Hersteller ein Produkt liefert, dass vom Endanwender oder von Ihnen stilisiert wird, dann sollten die Vorgaben auch validiert sein. Daher müsste der Hersteller ein Is-Produkt liefern. Falls Sie jedoch die Vorgaben selbst validieren müssen (gewissermaßen eigenständig sterilisieren) und sicherstellen müssen, dass das Produkt anschließend seine Funktion erfüllt, würden Sie quasi zum Hersteller werden. In diesem Fall müssten Sie ein vollständiges Konformitätsbewertungsverfahren durchlaufen (siehe auch Artikel 17 für Einmalprodukte), was wahrscheinlich ausgeschlossen wäre. Artikel 16 Absatz 1c) besagt ebenfalls, dass Sie zum Hersteller werden, wenn Sie etwas ändern, das einen Einfluss auf die Konformität hat. Die beste Lösung wäre wahrscheinlich, dass der Hersteller Sie mit der Sterilisation beauftragt und das Produkt bereits als Is kennzeichnet.
Ich hoffe, ich konnte helfen. Andernfalls fassen Sie gern nach oder kontaktieren Sie mich auch gern direkt.
Liebe Grüsse, Mario Klessascheck
Hallo liebes Johner-Team,
im Aritkel schreibt ihr, dass die MDR die Kombination eines Medizinprodukts mit einem Nicht-Medizinprodukt in einem System auch ohne erneute Konformitätsbewertung erfolgen kann, und argumentiert basierend auf der englischen Übersetzung (die ja den definierten Begriff *device* verwendet) , dass die MDR in Artikel 22(4) nur Medizinprodukte ohne CE-Kennzeichnung meint. Das ist vom Wortlaut her auch nachvollziehbar.
Nun verlangt die MDCG 2023-4 aber dass in Bezug auf die Verbindung von Medizinischer Software und Hardware in einem System, dass wenn *the hardware or hardware component is an integral part of a general consumer product or wearable digital product and is not a medical device or an accessory to a medical
device and has no intended medical purpose*, dann muss der Hersteller *comply with the requirements under equivalent conditions to a situation where a manufacturer is combining a medical device with another
product according to Article 22(4)*.
Damit sind laut MDCG 2023-4 *devices which do not bear the CE marking* sowohl bestimmte Nicht-Medizinprodukte (nämlich solche, die integraler Bestandteil eines Konsumentenproduktes ist) als auch Medizinprodukte ohne CE-Kennzeichung. DIe Folge ist, dass ein System aus Nicht-Medizinprodukten und Medizinprodukten als eigenständiges Medizinprodukt zertifiziert werden muss.
Ich sollte anmerken, dass In Fussnote 2 die MDCG 2023-4 angibt, dass *for the purposes of this document, hardware should not be understood to include desktop PC or cloud computing platform (server), und immer noch die Zulassung von nicht-medizinischen Geräten als Komponente, Zubehör oder eigenständiges Medizinprodukt zulässt. Für einfache Systeme blieben einem dann aber nur noch diese beiden nicht-medizinischen Gerätetypen.
Wie seht ihr das?
Lieber Herr Guggenberger,
sie haben absolut recht und ich teile Ihre Interpretation. Selbst für einen PC kann eine Konformitätsbewertung notwendig werden, wenn ein Patient über einen Sensor verbunden wird. Dann muss zusätzlich die elektrische Sicherheit für den Patienten sichergestellt werden. Ich werde das MDCG Dokument im Artikel ergänzen.
Nochmals vielen Dank für die sehr wertvolle Ergänzung!
Liebe Grüsse, Mario Klessascheck
Hallo zusammen,
wir stellen Behandlungseinheiten zusammen und vertreiben sie bislang nur innerhalb Deutschlands. Nun möchten wir diese auch nach Österreich versenden. Ist das als deutschsprachiges Land ohne inhaltliche Änderungen machbar? Genügt die Anmeldung beim BfArM und in EUDAMED? Und müssen die enthaltenen MDs/IVDs einzeln geprüft werden oder genügt es, dass alle Produkte CEs besitzen?
Viele Grüße
Marek Deckena
Lieber Herr Deckena,
Innerhalb der EU gelten für jedes Land die gleichen Bestimmungen. Es gibt Ausnahmen bei den Sprachen, natürlich nicht zwischen DE und A. Ob Sie die Produkte einzeln oder zusammen prüfen müssen, ist hingegen nicht länderspezifisch, sondern allgemeingültig. Sie dürfen die Produkte nur im Rahmen ihrer Zweckbestimmung miteinander kombinieren. Artikel 22 Absatz sagt, dass die gegenseitige Vereinbarkeit der Medizin- und gegebenenfalls sonstigen Produkte prüfen müssen.
Hilft Ihnen das weiter? Sonst haken Sie gern nach.
Liebe Grüsse, Mario Klessascheck
Guten Tag,
ich finde dieses Thema mit Systemen und Behandlungseinheiten irgendwie ganz schön kompliziert und bin mir nun überhaupt nicht sicher ob wir unsere Produkte danach eingruppieren müssen.
Wir sind Hersteller und verkaufen z.B. Otoskope (nur Griff & Kopf), Otoskope mit Ladebatterie, Otoskope mit Ohrtrichtern, Otoskop-Sets (mit Ohrtrichtern und verschiedenen Aufsätzen) und Otoskop-/ Ophthalmoskop-Sets (1 Griff + 1 Otoskop-Kopf und 1 Ophthalmoskop-Kopf).
Sind das nun alles Systeme und/oder Behandlungseinheiten?
Und falls ja, müssen die dann alle einzelne Konformitätserklärungen bekommen?
Ich hoffe Sie können mir weiterhelfen.
Vielen Dank im Voraus.
Liebe Frau Lerch,
Sie haben recht, die Abgrenzung ist nicht immer einfach. Eine Abgrenzung wäre wie folgt: Wenn Sie die Komponenten so verpacken, dass ein Anwender damit einen spezifischen medizinischen Zweck durchführen kann, handelt es sich um eine Behandlungseinheit. Hierbei liegt die Betonung auf den Wörtern „spezifisch“ und „verwenden“. Ein Beispiel wäre, sterile Handschuhe zusammen mit einem Blutentnahmeset zu verpacken, damit sie gemeinsam verwendet werden können. Bei einem System liegt die Betonung auf dem Teil der Definition: „verbunden oder kombiniert“, „um einen spezifischen medizinischen Zweck zu erfüllen“. Die Komponenten Ihres Otoskops sind dafür vorgesehen, miteinander verbunden zu werden, um den vorgesehenen Zweck zu erfüllen.
Ihre Produkte:
Da Sie die Rechtskonformität der einzelnen Komponenten nachweisen können, müssen Sie für die beiden Sets selbst keine zusätzliche CE-Kennzeichnung tragen. Es muss jedoch Ihren Namen, den eingetragenen Handelsnamen oder die eingetragene Handelsmarke sowie die Anschrift, unter der Ihre Firma kontaktiert werden kann, tragen.
Konnte ich damit helfen?
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck,
vielen Dank für Ihre ausführliche Antwort.
Sie haben mir wirklich sehr geholfen und ich glaube ich verstehe jetzt deutlich besser wann es sich um ein einzelnes Medizinprodukt, ein Sytsem oder eine Behandlungseinheit handelt.
Viele Grüße und einen schönen Tag.
d) Abgrenzungen zu Kombinationsprodukten
Kombinationen von Medizinprodukten und Arzneimitteln oder biologischen Produkten zählen als „Kombinationsprodukte“ und fallen nicht unter die Definition von „Systeme und Behandlungseinheiten
Wie lässt sich das belegen?
Meine Vermutung: Art.22 ist für ‚devices‘, von denen eins ein CE-Mark hat, Art 1(8) und Art 117 beschreiben Kombinationsprodukte. Gibt es einen Beleg, eine Stellungnahme (z.B. Team Notified Body oder EMA?)
Sehr geehrter Herr Postert,
Ihre Annahme ist korrekt. Kombinationsprodukte zählen nicht als Systeme und Behandlungseinheiten.
Es geht auch nicht um ein Entweder-Oder: Wenn es ein Kombinationsprodukt ist, dann müssen die gesetzlichen Anforderungen daran erfüllt werden. Diese Anforderungen an Kombinationsprodukte gehen weit über die Anforderungen hinaus, welche die MDR and Systeme und Behandlungseinheiten stellt. Details finden Sie im Fachartikel zu den Kombinationsprodukten.
Beste Grüße, Christian Johner
Hallo liebes Johner-Team,
fällt ein Dermatoskop, welches eine App zur Speicherung der Daten besitzt und auf einem Endgerät läuft, dass nicht zum Produktumfang gehört, unter die Definition des „Systems“ sowohl im Kontext der MDR als auch der IEC 60601-1?
Vielen Dank im Voraus für Ihre Unterstützung!
Liebe Frau Stolte,
Wenn Sie das Endgerät nicht vertreiben, müssen Sie keine Artikel 22 Erklärung ausstellen. Aus Sicht der IEC 60601-1 wäre es als System zu betrachten, wenn Sie die Nutzung mit dem Endgerät so vorgesehen haben. Wenn es eine elektrische Verbindung gibt, müssen Sie die Qualität der Isolation bei der Safety Prüfung berücksichtigen. Bei der EMV Prüfung sollten Sie stellvertretend ein Endgerät mit testen, oder zumindest das Kabel als SIP/SOP mit prüfen lassen.
Beste Grüsse, Mario Klessascheck
Hallo Herr Klessascheck,
ich benötige erneut Ihre Fachexpertise. Wir sind Inverkehrbringer von sterilen Behandlungseinheiten nach Art. 22,3. Aktuell sind wir gem. 93/42/EWG Anhang V zertifiziert und haben den Antrag bei unserer benannten Stelle gestellt. Gem. Art. 22,3 steht es uns frei, welches Konformitätsbewertungsverfahren wir wählen. Wir haben uns für das Verfahren gem. Anhang IX I und III entschieden. Nun haben wir die Antragsprüfung zurück erhalten mit folgendem NC: In unserem QMH haben wir unter Abschnitt 7.4 Ausschlüsse und NIcht Anwendungen 7.3 Entwicklung ausgeschlossen. Und es sei jetzt nicht nachvollziehbar, wie ein Verfahren nach Anhang IX Kapitel I und II durchgeführt werden soll. Können Sie vielleicht Licht ins Dunkle bringen? Ich sehe darin keinen Widerspruch. Gem. Art 22,3 steht es mir frei zu wählen, wobei sich die Anwendung dieser Verfahren und die Beteiligung der benannten Stelle nur auf die Apsekte des Sterilisationsverfahrens beschränkt. Das bedeutet doch, dass wir lediglich die Validierungen für Verpackung, Transport und Sterilisation einreichen müssen. Ist dies korrekt? Wie wird denn aber Art. 22 geprüft, sprich dass nur CE markierte Produkte verwendet werden?
Liebe Frau Kuhlmann,
Ohne die genauen Begründungen für die NCs zu kennen, ist es nicht einfach, die Situation vollständig zu beurteilen, aber ich werde es dennoch versuchen. Zunächst sollten Sie kein Kapitel aus Anhang IX ausschließen. Dies könnte ein erstes Missverständnis sein. Wenn die Ausschlüsse im Widerspruch zum festgelegten Geltungsbereich Ihres aktuellen QM-Systems stehen, kann dies bereits zu Nebenabweichungen oder sogar Hauptabweichungen führen. Es ist schwierig, den gesamten Normpunkt 7.3 auszuschließen, da es wahrscheinlich Anforderungen gibt, die möglicherweise unter ISO 13485, Punkt 7.3.3 (Entwicklungseingaben) fallen.
Wir empfehlen, alle Normpunkte unter 7.3 im Detail zu prüfen und für jede Nichtanwendung eine fundierte Begründung zu formulieren. So kann genauer geprüft werden, ob eine Entwicklungsplanung (Punkt 7.3.2) wirklich ausgeschlossen werden kann.
Zudem sollten Sie beachten, dass über den gesamten Produktlebenszyklus hinweg ein Risikomanagement gefordert wird, das ebenfalls Bestandteil der Technischen Dokumentation (TD) ist.
Hilft Ihnen das weiter?
Liebe Grüsse, Mario Klessascheck
Hallo liebes Johner-Team,
fällt ein Sirup mit Messbecher, wobei der Messbecher bereits nach MDR-zertifiziert ist und bei jedem Sirup mit dabei ist, unter die Definition einer Behandlungseinheit oder würde man hier trotzdem von einem Medizinprodukt (Sirup) mit Zubehör im Kontext der MDR ausgehen?
Vielen Dank im Voraus für Ihre Unterstützung!
Liebe Frau Tabaka,
Wenn der Sirup ein Medizinprodukt ist, dann können Sie den Messbecher zusammen mit dem Sirup als System vertreiben. Das wäre auch Artikel 22. Der Begriff Zubehör ist nur aus Sicht des Herstellers des Messbechers relevant. Aus Ihrer Sicht ist der Messbecher ein Medizinprodukt. Sie verpacken somit zwei Medizinprodukte, die für den gemeinsamen Gebrauch bestimmt sind und hätten damit ein System. Sie müssten wahrscheinlich noch die Verträglichkeit des Sirups mit dem Material des Messbechers betrachten. Aber das würden Sie schon in der Produktakte des Sirups machen, da der Sirup dafür bestimmt ist, mit einem Messbecher eingenommen zu werden.
Hilft Ihnen das weiter?
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Mario Klessascheck,
vielen Dank für ihre Antwort! Ja, das hat uns einen Schritt weitergebracht, vielen lieben Dank!
Sehr geehrter Herr Klessascheck,
wir sind Hersteller von Arthroskopiepumpen und Schlauchsets (Einweg- und wiederverwendbare Schläuche) zur Spülung und Absaugung für die Einbringung und Ableitung von Flüssigkeit, die die Kavität für minimalinvasive Eingriffe bildet.
In der Vergangenheit (unten MDD) haben wir die Kombination Pumpe – Schlauchsatz als System abgegrenzt und einige TD-Aufzeichnungen (Risikomanagement, Gebrauchsanweisung, Klinische Bewertung, PMS-Dokumentation) für beide Technischen Dokumentationen erstellt.
Jedes Medizinprodukt oder Zubehör für ein Medizinprodukt des Systems muss jedoch eine eigenständige Technischen Dokumentationen haben, die auch die Erfüllung der grundlegenden Anforderungen nachweisen kann.
Wir haben diese Abgrenzung auch unter MDR gemacht und wurde von der benannten Stelle abgelehnt.
Die Begründung war: „Die Kombination von Medizinprodukten kann als Systemabgegrenzt werden, wenn eine 1 zu 1 Verbindung der Schnittstellen und eine direkte und kontinuierliche Verbindung der Medizinprodukte gegeben ist.“
Die Schlauchsätze werden nach einer Anwendung entweder weggeworfen oder aufbereitet und somit ist die kontinuierliche Verbindung nicht gegeben. Somit haben wir nicht mehr die Möglichkeit die obengenannten Dokumente für beide Technischen Dokumentationen zu erstellt.
Wie bewerten Sie die Auslegung der benannten Stelle zum Thema System Gerät-Schlauchsatz?
Ich bedanke mich vorab für Ihre Rückmeldung!
Lieber Herr Adeil,
Im Grunde genommen hat die benannte Stelle recht. Ich betrachte das Schlauchset als Zubehör und damit als eigenständiges Produkt mit eigener technischer Dokumentation. Da Sie sowohl die Pumpe als auch das Schlauchset selbst herstellen, sehe ich keinen Vorteil darin, diese als System zu deklarieren. Das Schlauchset kann unter der Maßgabe vertrieben werden, dass es ausschließlich mit Ihrer Pumpe verwendet wird. Das Zusammenspiel beider Komponenten wurde bereits auf Produktebene geprüft – eine zusätzliche Erklärung nach Artikel 22 halte ich daher nicht für notwendig. Sollten Schlauchset und Pumpe nach der Anwendung gemeinsam entsorgt werden, könnten Sie die Kombination theoretisch als Behandlungseinheit gemäß Artikel 22 deklarieren. Auch darin sehe ich jedoch keinen nennenswerten Vorteil.
Vielleicht schildern Sie mir nochmals kurz, was Sie durch die Systemdeklaration erreichen möchten (gern auch direkt per Email).
Liebe Grüsse, Mario Klessascheck
Guten Tag
Ich habe eine Frage im Zusammenhang mit der Konformitätserklärung gemäß der Medizinprodukteverordnung (MDR) sowie der Erklärung nach Artikel 22.
Wir sind als Händler tätig und beabsichtigen, eine Behandlungseinheit von einem Lieferanten zu beziehen. Diese Einheit besteht aus mehreren Produkten, die sowohl einzeln als auch in Kombination verwendet werden können. Um die Behandlungseinheit rechtmäßig vertreiben zu dürfen, müssen wir sicherstellen, dass alle enthaltenen Produkte den Anforderungen der MDR entsprechen.
Unserer Auffassung nach ist es erforderlich, dass der Lieferant uns die Konformitätserklärungen für jedes einzelne Produkt innerhalb der Einheit zur Verfügung stellt. Der Lieferant hingegen ist der Ansicht, dass eine Erklärung gemäß Artikel 22 ausreichend sei, um die Konformität aller Produkte abzudecken. Wichtig zu erwähnen ist hier noch, dass unser Lieferant die Produkte zum Teil selbst herstellt und zum Teil von anderen Herstellern einkauft.
Da der entsprechende Abschnitt in der MDR für uns nicht eindeutig ist, möchte ich mich auf diesem Weg erkundigen, wie dies korrekt zu handhaben ist.
Vielen Dank für Ihre Unterstützung.
Freundliche Grüße
Liebe Frau Müller,
Für Sie als Händler sind die Vorgaben in Artikel 14 der MDR maßgeblich.
Sie sind dabei nicht verpflichtet zu prüfen, ob die einzelnen Komponenten die Anforderungen der MDR erfüllen. Stattdessen müssen Sie sicherstellen, dass:
• eine CE-Kennzeichnung vorhanden ist,
• eine EU-Konformitätserklärung (in Ihrem Fall eine Erklärung nach Artikel 22) vorliegt,
• die Kennzeichnung korrekt ist und
• eine Gebrauchsanleitung beiliegt.
Sollten Sie den Verdacht haben, dass die Produkte nicht den Vorgaben der MDR entsprechen, dürfen Sie diese nicht in Verkehr bringen. In einem solchen Fall sollten Sie den Hersteller, den Bevollmächtigten oder den Importeur informieren. Eine Konformitätserklärung ist kein geheimes Dokument. Wenn sich der Lieferant nicht kooperativ zeigt, kann das Indiz dafür sein, dass Sie Ihre weiteren Pflichten nur mit erheblichem Aufwand oder gar nicht erfüllen können und vielleicht von einem Vertrieb absehen sollten.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Klessascheck
Besten Dank führ Ihre Antwort!
Darf ich fragen, wie es aussieht, wenn ich EC-REP dieses Kits wäre. Würde dann ebenfalls die Erklärung nach Artikel 22 ausreichen oder bräuchte ich hier die Konformitätserklärungen der einzelnen Produkte?
Freundliche Grüsse