
Um die Biokompatibilität von Standardmaterialien nachzuweisen, können Medizinproduktehersteller in den meisten Fällen auf Tierversuche verzichten.
Dem Johner Institut ist es in den letzten Jahren immer gelungen, Alternativen zu Tierversuchen aufzuzeigen, selbst wenn Behörden und Benannte Stellen diese forderten.
Erfahren Sie hier, wie Sie einen Beitrag zum Tierwohl leisten und dabei sowohl Geld sparen als auch Ihre Medizinprodukte schneller in den Markt bringen können.
1. Über Tierversuche
a) Nachweise, für die sich Tierversuche eignen
Hersteller müssen die Sicherheit, Leistungsfähigkeit und Wirksamkeit von Medizinprodukten nachweisen. Tierversuche sind eine Möglichkeit, um diese Nachweise zu führen:
| Nachzuweisende Eigenschaft | Beispiel |
| Biologische Sicherheit, Biokompatibilität | Tiere werden in Kontakt mit einem Material/Extrakt des Medizinprodukts gebracht, um nachzuweisen, dass es keine biologischen Reaktionen zeigt. |
| Leistungsfähigkeit | Tieren werden neu entwickelte orthopädische oder kardiologische Implantate eingesetzt, um die Leistung zu bewerten. |
| Wirksamkeit | Tiere mit Leberversagen werden an ein Gerät zur Leberdialyse angeschlossen, um nachzuweisen, dass es die leberpflichtigen Substanzen ausreichend entfernt und die Überlebensrate der Tiere erhöht. |
Diese Tabelle sagt nicht aus, dass die nachzuweisenden Eigenschaften nur mit Tierversuchen erbracht werden können.
b) Nachweis der Biokompatibilität
Dieser Artikel konzentriert sich auf den Nachweis der biologischen Sicherheit bzw. Biokompatibilität, d. h. der Verträglichkeit von Materialien des Produkts, die direkt oder indirekt (z. B. über Flüssigkeiten oder Gas) in Kontakt mit dem menschlichen Körper kommen.
In diesem Artikel zur Biokompatibilität erfahren Sie, weshalb Materialzertifikate nicht immer ausreichen.
Hersteller müssen dabei nachweisen, dass keine Substanzen in Mengen aus dem Material heraus- oder von der Oberfläche heruntergelöst werden (z. B. Rückstände aus Produktion/Reinigung), die toxikologisch relevant sind, also gesundheitsgefährdende Effekte haben.
Es können unterschiedliche gesundheitsschädliche Effekte je nach Art und Dauer der Anwendung des Medizinprodukts auftreten. Diese Effekte werden klinische Endpunkte genannt. Man kann zwischen lokalen Endpunkten, also anwendungsortsbezogener Effekte und systemischen Endpunkten, die den ganzen Organismus betreffen, differenzieren.
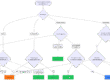
Für die meisten Endpunkte können für die Bewertung Tierversuche herangezogen werden (s. Tab. 2). Je nach Anwendung und verwendeten Materialien muss die Biokompatibilität jedoch nicht mittels Tierversuchen bewertet werden. Es gibt geeignete Möglichkeiten, die Endpunkte auch ohne Tierversuche zu adressieren.
| Endpunkte für die biologische Bewertung | Systemischer Endpunkt? | Ist das TTC-Konzept* dafür vorgesehen? | Kann der Endpunkt mit Tierversuchen nachgewiesen werden? | Muss der Endpunkt mit Tierversuchen nachgewiesen werden? |
| Physikalische und/oder chemische Informationen (Materialeigenschaften) | Nein | Nein | Nein | Nein |
| Zytotoxizität (Zellschädigung) | Nein | Nein | Nein | Nein |
| Sensibilisierung der Haut (z. B. allergische Reaktion) | Nein | Nein | Ja | Nein |
| Irritation der Haut oder des Gewebes (z. B. Rötung und Juckreiz) | Nein | Nein | Ja | Nein |
| Systemische Toxizität: akute, subakute, subchronische und chronische Toxizität (z. B. Organschäden, Tod) | Ja | Ja | Ja | Nein |
| Materialbedingte Pyrogenität (Erzeugen von Fieber) | Ja | Nein | Ja | Nein |
| Genotoxizität (Schädigung der DNA) | Ja | Ja | Ja | Nein |
| Karzinogenität (Verursachen von Krebs) | Ja | Ja | Ja | Nein |
| Reproduktionstoxizität (Beeinträchtigung der Fortpflanzungsfähigkeit) | Ja | Ja | ja | Nein |
| Implantationseffekte (z. B. Abstoßung von Implantaten) | Nein (lokale Effekte) Ja (systemische Effekte) | Ja (systemische Effekte) | Ja (gemeinsame Betrachtung möglich) | Meistens |
| Blutverträglichkeit (z. B. Thrombose) | Nein | Nein, aber In-vitro-Tests | Ja | Nein (außer Implantate) |
* TTC-Konzept (Threshold of Toxicological Concern): s. Kap. 3
Ablauf von Tierversuchen
Zum Nachweis der Biokompatibilität werden die Tiere (z. B. deren Haut, Augen, Blut) in direkten Kontakt mit dem zu prüfenden Material bzw. einem Extrakt davon gebracht. Dazu sind mehrere Tiere (üblicherweise mindestens fünf pro Endpunkt, Dosis und Aufnahmeweg) notwendig.
Für notwendige Konzentrationsbestimmungen werden Tiere Vorversuchen ausgesetzt. Für die Prüfung selbst werden zusätzliche Tiere für die Kontrollgruppen gebraucht, um die Eignung des Prüfsystems zu zeigen.
Als Versuchstiere dienen vorwiegend Meerschweinchen, Kaninchen, Ratten oder Mäuse. Am Ende der Prüfung werden diese schmerzfrei getötet, um weitere Untersuchungen für die Bewertung des Endpunkts durchführen zu können.
Aufwand und Kosten von Tierversuchen
Tierversuche dauern typischerweise einige Wochen. Manchmal verzögern sie die Zulassung um mehrere Monate. Dies ist insbesondere der Fall, wenn
- der Hersteller viele Endpunkte zu bestimmen hat (es kann meistens nur ein Endpunkt und Aufnahmeweg pro Versuch untersucht werden),
- der Versuchsaufbau erst noch zu entwickeln und validieren ist,
- die Ergebnisse nahelegen, dass die Biokompatibilität nicht gegeben ist sowie
- bei nicht eindeutigen Ergebnissen und produktunabhängigen Einflüssen.
Die Kosten hängen stark von Art und Anzahl der Endpunkte ab. Sie belaufen sich fast immer auf fünfstellige Summen. Beispielsweise dauert die Bestimmung eines einzigen Endpunkts (z. B. ein Sensibilisierungstest) in der Regel 14 bis 16 Wochen. Die Kosten dafür liegen ca. zwischen 5.000 und 10.000 EUR.
2. Regulatorischer Rahmen
Eine gesetzliche oder normative Pflicht zu Tierversuchen besteht nicht.
Die Normen zur Biokompatibilität (Normenreihe ISO 10993) verlangen „nur“ eine adäquate Bewertung der Biokompatibilität des finalen Endprodukts.
Die ISO 10993-1:2025 fordert eindeutig ein Vorgehen, dass zunächst eine Materialcharakterisierung und In-vitro-Tests erfolgen sollen und nur auf Tierversuche ausgewichen werden darf, falls die gewonnenen Daten nicht ausreichen.
Any in vivo testing shall be conducted in accordance with ISO 10993-2. The conduct of any in vivo biological testing shall be minimised to that which is strictly necessary to complete the biological evaluation. In the following order, the biological evaluation shall where possible:
ISO 10993-1:2025, 4.4 Animal welfare
— refine biological risk estimation by considering additional information in the literature or conducting additional data analysis, or by generating additional chemical and physical information (see 6.8.2);
— replace the use of animal testing with alternative techniques (e.g. in vitro or in silico) where these methods provide equally relevant information to that obtained from in vivo models;
— reduce the number of animals used to a minimum to obtain information from fewer animals or to maximize the amount of data collected from each animal in a way that is compatible with the study objectives; and
— refine the way experiments are carried out to minimize animal pain and distress.
The approach taken shall be justified and documented.
Ergeben sich aus der Materialcharakterisierung keine Hinweise auf freigesetzte Stoffe mit toxikologisch relevantem Potenzial, kann der Verzicht auf zusätzliche Prüfungen nicht nur gerechtfertigt, sondern sogar verpflichtend sein.
Die FDA fordert ebenfalls einen „3R-Ansatz“ bezüglich der Durchführung von Tierversuchen: „reduce, refine and replace“. Die FDA hat in diesem Zusammenhang in der aktuell überarbeiteten Version der Guidance „Use of International Standard ISO 10993-1, „Biological evaluation of medical devices – Part 1: Evaluation and testing within a risik management process“ (Sept.2023) den Anhang G „Biocompatibility of Certain Devices in Contact with Intact Skin“ veröffentlicht. Es werden Produktmaterialien mit geringem Risiko für den Kontakt mit intakten Hautoberflächen identifiziert, die seit Jahren sicher in Medizinprodukten eingesetzt werden, sowie ein Vorgehen beschrieben, um auf Prüfungen/Tierversuche zu verzichten. Hierzu werden Anforderungen an das Qualitätssystem und andere Kontrollen aufgeführt, um Biokompatibilitätsprobleme bei Medizinprodukten, die mit intakter Haut in Berührung kommen, umgehend zu erkennen und zu vermeiden.

”FDA believes that these materials pose a very low biocompatibility risk because they have a long history of safe use in legally marketed medical devices that contact intact skin. The policy outlined in this Attachment describes a least burdensome76approach for these devices that recommends specific material information to be included in a premarket submission in lieu of biocompatibility testing. This approach also supports the principles of the “3Rs,” to replace, reduce, and/or refine animal use in testing when feasible”
Guidance-Dokument der FDA zur Anwendung der ISO 10993-1:2023
Außerdem ist im Guidance-Dokument der FDA im Allgemeinen ein Vorgehen gefordert, welches Tierversuche bei der Bewertung der Biokompatibilität von Medizinprodukten aus Standardmaterialien vermeiden oder reduzieren soll.
“Some devices are made of materials that have been well characterized both chemically and physically in the published literature and/or have a long history of safe use in legally marketed medical devices. It may not be necessary to conduct testing for all or a portion of the biocompatibility endpoints suggested in the FDA matrix of this guidance.“
Guidance-Dokument der FDA zur Anwendung der ISO 10993-1:2023
“For FDA submissions, biocompatibility information for the device in its final finished form, either developed through the risk management process or from biocompatibility testing (using both in vitro and in vivo models), and/or adequate chemical, physical, morphological, and topographical characterization in conjunction with supplementary biocompatibility information that adequately address the biocompatibility risks of the device should be provided.” (FDA)
Guidance-Dokument der FDA zur Anwendung der ISO 10993-1:2023
In der chemischen Charakterisierung findet dann das sogenannte TTC-Konzept Anwendung, um die Biokompatibilität der Medizinprodukte und somit die Patientensicherheit zu gewährleisten.
3. Das TTC-Konzept: Threshold of Toxicological Concern
a) Ziel des TTC-Konzepts
Das TTC-Konzept wurde entwickelt, um das Risiko durch Substanzen, die in kleinen Mengen vorkommen, qualitativ zu bewerten [EFSA].
Das Ziel des Konzepts ist es, ein Expositionsniveau für alle Chemikalien festzulegen, unabhängig davon, ob es chemikalienspezifische Toxizitätsdaten gibt oder nicht, unterhalb dessen kein nennenswertes Risiko für die menschliche Gesundheit besteht.
b) Festlegung des TTC
Der TTC ist damit ein Schwellenwert für toxikologische Bedenken und wird in der Regel für Chemikalien mit unbekannter Toxizität festgelegt, unterhalb dessen die Wahrscheinlichkeit schädlicher Auswirkungen auf die menschliche Gesundheit aufgrund der chemischen Struktur und geringen Dosis vernachlässigbar ist.
Dieses Konzept basiert auf der Annahme, dass eine tägliche Exposition von 1,5 µg einer unbekannten organischen Verbindung mit einem akzeptablen Krebsrisiko von unter 1:1.000.000 über die gesamte Lebensdauer verbunden ist.
Das bedeutet demnach, dass Expositionen, die kürzer als lebenslang sind, mit einer proportional höheren täglichen Aufnahme möglich sind. Der TTC bei einer Anwendung von weniger als 10 Jahren beträgt demnach 10 µg/Tag, bei Kurzzeitanwendungen von weniger als einem Monat 120 µg/Tag.
c) Verwendung des TTC
Auf Basis des ermittelten TTC wird eine analytische Bewertungsschwelle (AET = Analytical Evaluation Threshold) errechnet, von der die benötigte Bestimmungsgrenze der chemischen Analytik abhängt. Mit dem AET-Wert wird sichergestellt, dass die Analysen ausreichend empfindlich durchgeführt werden. In die Berechnung des AET fließen zusätzlich zum TTC weitere Parameter wie die Anzahl eingesetzter Produkte, das Extraktionsvolumen, Patientengruppen, der Unsicherheitsfaktor und die Anwendung (Anzahl eingesetzter Produkte pro Patient und Tag) ein.
Das TTC Konzept ermöglicht eine wertvolle Alternative zu Tierversuchen. Grundsätzlich wurde dieser Ansatz zwar für systemische Endpunkte entwickelt. Dieser kann aber auch zur Bewertung von lokalen Endpunkten wie Irritation und Sensibilisierung von Standardmaterialien zur Bewertung des finalen Endprodukts herangezogen werden.
So beschreibt die ISO 10993-1 zum einen, dass eine chemische Charakterisierung nach ISO 10993-18 mit geeignetem TTC-Konzept ausreicht, um festzustellen, ob noch weitere Prüfungen notwendig sind. Zum anderen fordert z. B. die ISO 10993-10 zur Prüfung auf Hautsensibilisierung ein stufenweises Vorgehen mithilfe von Materialcharakterisierung und chemischer Analyse auch für die Bewertung dieses Endpunkts.
ISO 10993-10: “Dieses Dokument fordert ein stufenweises Vorgehen, das einen oder mehrere der folgenden Schritte enthalten muss:
ISO 10993-10:2021, 4
a) Charakterisierung des Prüfmaterials, die eine chemische Charakterisierung und Analyse des Prüfmusters nach den allgemeinen Grundsätzen, wie in ISO 10993-9, ISO 10993-13, ISO 10993-14, ISO 10993-15, ISO 10993-17 und ISO 10993-18 beschrieben, einschließt;
b) Literaturstudie, die eine Bewertung der chemischen und physikalischen Eigenschaften und die Gewinnung von Informationen über das Sensibilisierungspotential aller Bestandteile des Produkts sowie über strukturell nahe stehende Chemikalien und Materialien umfasst;
c) nach ISO 10993-2 muss die Bevorzugung von In-vitro-Prüfungen gegenüber In-vivo-Prüfungen beachtet werden und das Ersetzen letzterer, wenn neue In-vitro-Verfahren wissenschaftlich validiert und in vernünftiger Weise und praktisch verfügbar sind. Es gibt derzeit eine Reihe von international validierten und anerkannten In-vitro-Prüfungen zum Nachweis des Hautsensibilisierungspotentials von Chemikalien; keine dieser Prüfungen ist jedoch als geeignet für die Verwendung mit Medizinprodukten ausgewiesen.“
Vorhandene Daten aus der Literatur über bereits durchgeführte Tierversuche zum Material selbst, zu den verwendeten Additiven sowie gefundenen Freisetzungsprodukten können zur Bewertung der Exposition herangezogen werden.
4. Verzicht auf Tierversuche
a) Wann ein Verzicht notwendig ist
Bei Standardmaterialien ist der Nachweis der Biokompatibilität über Materialcharakterisierung, In-vitro-Tests und chemische Charakterisierung mittels TTC-Konzept nicht nur möglich, sondern sogar gefordert.
b) Wann ein Verzicht möglich ist
Generell ist ein Verzicht auf Tierversuche bei allen o. g. Endpunkten möglich (s. Tab. 2, rechte Spalte), insbesondere bei den Endpunkten, bei denen das TTC-Konzept anstandslos anwendbar ist (s. Tab. 2, mittlere Spalte).
Dem Johner Institut ist es in den letzten Jahren immer gelungen, eine Argumentationslinie für den Verzicht auf Tierversuche zu finden, selbst wenn Behörden oder Benannte Stellen Tierversuche explizit gefordert haben.
c) Wann ein Verzicht nicht möglich ist
Wenn keine Literaturdaten und Erfahrungen zum Material bzw. Produkt vorliegen und auch nicht abgeleitet werden können, sind Tierversuche notwendig. Das ist v. a. bei neuartigen Materialien der Fall.
d) Vorteile des Verzichts auf Tierversuche
Die Strategie über In-vitro-Tests hat im Vergleich zu Tierversuchen viele Vorteile:
- Das Tierwohl wird nicht beeinträchtigt.
- Die Tests sind sensitiver.
- Die Tests sind besser reproduzierbar.
- Mehrere Endpunkte und Aufnahmewege (oral/dermal/inhalativ) sind bewertbar.
- Die Tests sind (deshalb) meist kostengünstiger und weniger zeitintensiv.
- Die Tests erlauben eine bessere Vergleichbarkeit von Produkten.
- Ergebnisse können ggf. zur Ursachenanalyse auf Materialien übertragen werden.
Im Gespräch mit Professor Johner berichtet unsere Leiterin des Bereichs „Biocompatibility“, was bei der Biokompatibilität nachzuweisen ist und wie es dabei gelingt, (fast) immer auf Tierversuche zu verzichten.
Diese und weitere Podcast-Episoden finden Sie auch hier.
5. Vorgehen
Um auf Tierversuche verzichten zu können, sollten Sie als Hersteller die folgenden Schritte gehen:
- Erstellen Sie einen Biological Evaluation Plan (BEP) individuell für das Medizinprodukt. Darin müssen Sie Ihre Strategie zur Bewertung der Biokompatibilität ohne Tierversuche rechtfertigen und das Vorgehen beschreiben. Außerdem sind die Testparameter sowie die jeweiligen Grenzwerte spezifisch für das Medizinprodukt festzulegen, und zwar abhängig von der Exposition (z. B. über das TTC-Konzept).
- Das Johner Institut empfiehlt bei Unsicherheiten, diesen BEP mit den Behörden bzw. Benannten Stellen abzustimmen.
- Suchen Sie ein geeignetes Labor, welches auf die notwendigen Prüfungen spezialisiert ist. Vorzuziehen sind akkreditierte/zertifizierte Labore mit entsprechender Erfahrung in dem jeweiligen Bereich.
- Fassen Sie die Ergebnisse des Labors in einem Biological Evaluation Report (BER) zusammen. Der BER enthält auch eine toxikologische Bewertung dieser Daten basierend auf den im BEP festgelegten Grenzwerten sowie Daten aus Literatur und Erfahrung.
6. Zusammenfassung und Fazit
Unnötige Tierversuche durchzuführen ist unethisch, teuer und langwierig.
In vielen Fällen sind Tierversuche sogar ungeeignet. Es ergibt wenig Sinn, bei Standardmaterialien wie einem Polyethylen-Gehäuse eines Bedienelements ein weiteres Mal durch einen Tierversuch zu prüfen, ob eine allergische Reaktion ausgelöst werden kann.
Zwar ist die Bewertung des finalen Endprodukts gefordert, aber nicht grundsätzlich über Tierversuche. Im Gegenteil:
Die ISO 10993-1 fordert, die Materialcharakterisierung als Methode vorzuziehen und Tierversuche zu vermeiden. Auch die FDA fordert in ihrem veröffentlichten Guidance-Dokument zum Thema Biokompatibilität von Medizinprodukten, Tierversuche zu reduzieren und durch geeignete In-vitro-Strategien zu ersetzen.
Das heißt: Behörden und Benannte Stellen können sich in diesen Fällen einer stringenten und datenbasierten Argumentationslinie nicht verweigern und müssen auf eine Forderung nach Tierversuchen verzichten.
So können Sie als Hersteller nicht nur zum Tierwohl beitragen, sondern gleichzeitig Aufwände, Kosten und Zeit sparen.
Änderungshistorie
- 2025-12-17: Anpassung: Zitat aus der aktuellen ISO 10993-1:2025
- 2023-09-14: Zitate und Verlinkung auf aktuelle FDA Guidance zur Anwendung der ISO 10993-1:2023 angepasst sowie Verweis auf den neuen Anhang G der Guidance eingefügt
- 2023-02-09: Erstellung des Blogbeitrags


Sie schreiben „Dem Johner Institut ist es in den letzten Jahren immer gelungen, Alternativen zu Tierversuchen aufzuzeigen“. Trift das für alle wichtigen Märkte zu? Letztlich würde es einem weltweit agierenden Hersteller wenig nutzen, die Behörden/NB in den USA und der EU zu überzeugen wenn die Behörde eines großen Marktes, z.B. in China die Tierversuche weiter verlangt.
Wie sind da Ihre Erfahrungen? Ist die Sicht der Behörden hier in den wichtigsten Märkten ähnlich?
Sehr geehrter Herr Müller,
die Aussage betrifft hauptsächlich die Märkte Europa und USA.
Bei China empfiehlt es sich tatsächlich die Strategie zusammen mit einem Partner vor Ort zu erarbeiten.
Herzliche Grüße