
Die Unterschiede zwischen der MDR und MDD sind groß. Dieser Artikel stellt diese Unterschiede vor. Damit verschafft er Herstellern, die ihre Produkte noch unter einer EU-Richtlinie (insbesondere MDD) in den Markt gebracht haben, einen Überblick über die zu schließenden „Gaps“.
1. Wie es zu den Unterschieden zwischen MDR und MDD kam
Die Medizinprodukte-Verordnungen (MDR, IVDR) lös(t)en die Medizinprodukte-Richtlinien (MDD, AIMD, IVD) ab.
Die neue EU-Verordnung zu Medizinprodukten (Medical Device Regulation, MDR, mit der Nummer 2017/745) ersetzt zwei Medizinprodukte-Richtlinien, nämlich die
- Richtlinie 93/42/EWG über Medizinprodukte (Medical Device Directive, MDD)
- Richtlinie 90/385/EWG über aktive implantierbare Medizinprodukte (Active Implantable Medical Devices, AIMD)
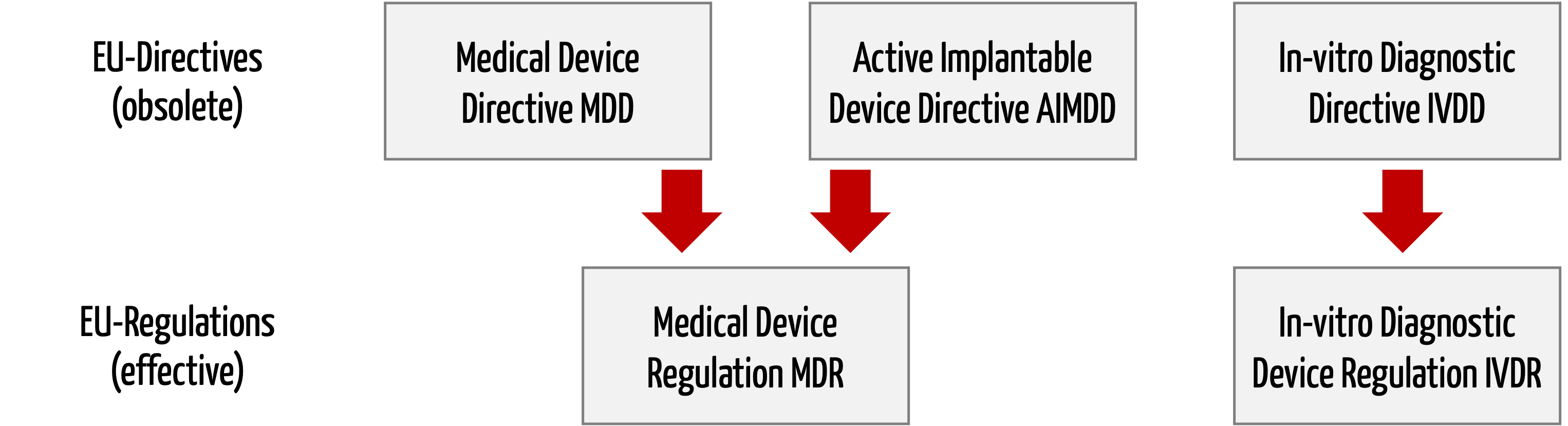
Die Richtlinie 98/79/EG über In-vitro Diagnostika (IVD) wird nicht in der Medical Device Regulation aufgehen, sondern durch eine eigene neue EU-Verordnung ersetzt (In-Vitro Diagnostic Medical Devices Regulation, IVDR, Nummer 2017/746).
Lesen Sie hier mehr zum Thema In-Vitro Diagnostic Medical Devices Regulation, IVDR.
2. Unterschiede bei den Produkteanforderungen
Die grundlegenden Anforderungen (jetzt grundlegende Sicherheits- und Leistungsanforderungen) im Anhang I der MDR sind deutlich umfangreicher als im Anhang I der MDD. Das betrifft auch die Anforderungen an die Begleitinformationen.
Die Anforderungen an den Inhalt der Technischen Dokumentation werden in einem neuen Anhang II der Medical Device Regulation deutlich detaillierter geregelt. Die kontinuierliche Aktualisierung dieser Unterlagen ist jetzt explizit gefordert. Lesen Sie weiter unten mehr zu den geänderten Anforderungen in der technischen Dokumentation.
Die MDR gibt umfangreiche Vorgaben für die Post-Market-Surveillance. Dazu zählen ein Post-Market-Surveillance-Plan ebenso wie Berichte (z. B. Periodic Safety Update Report, PSUR).
Klinische Bewertungen und klinische Prüfungen werden viel detaillierter geregelt. Dabei wird die Medical Device Regulation auch konkreter, was Art und Qualität der klinischen Daten betrifft. Die Daten aus der Post-Market Surveillance sind im Rahmen des Post-Market Clinical Follow-up (PMCF) einzubeziehen, um die klinische Bewertung regelmäßig zu aktualisieren.
Es gibt höhere Anforderungen an Produkte mit Gefahrstoffen, insbesondere an krebserzeugende, erbgutverändernde und fortpflanzungsgefährdende Stoffe.
Die EU-Kommission behält sich vor, sogenannte gemeinsame Spezifikationen (common specifications) festzulegen, wenn sie der Meinung ist, dass harmonisierte Normen fehlen oder unzureichend sind.
Jedes Produkt muss zukünftig eine eindeutige Produktidentifizierungsnummer (UDI) erhalten.
Die Anforderungen an die Wiederaufbereitung von Einmalprodukten sind gestiegen.
3. Unterschiede bei der technischen Dokumentation
Eine Übersicht über die Änderung der Anforderungen an die grundlegenden Anforderungen verschaffen die beiden folgenden Tabellen.
a) Vergleich: Produktbeschreibung und Labeling
| Kategorie | Detailforderung | MDR | MDD |
| Beschreibung des Geräts | Produkt- oder Handelsname
Allgemeine Beschreibung Klassifizierung und Begründung Varianten, Konfigurationen Fotos | X
X X X X | (X)
X (X) (X) X |
| Identifikation | UDI DI | X | – |
| Bestimmungs-gemäßer Gebrauch | Zweckbestimmung
Vorgesehene Anwender Patientenpopulation Indikationen und Kontraindikationen Vorgesehene Anwendung Zubehör Zusammenspiel mit anderen Produkten | X
X X X X X X | X
– – – (X) – (X) |
| Physikalisches Prinzip | Beschreibung der Neuerungen
Überblick über ähnliche Produkte Überblick über Vorgängerprodukte | X
X X | –
– – |
| Aufbau | Wesentliche Funktionen
Teile, Komponenten, Zusammensetzung Zeichnungen, Diagramme, Erläuterungen Beschreibung von Materialien (mit Körperkontakt) Technische Spezifikationen Berechnungen | X
X X X X – | –
X X – – X |
| Labeling | Broschüren, Kataloge
Gebrauchsanweisungen Beschriftungen Verpackungen | X
X X X | –
X X – |
b) Vergleich: Entwicklung, Produktion, nachgelagerte Phase
| Kategorie | Detailforderung | MDR | MDD |
| Herstellung | Entwicklungs- und Produktionsphasen inklusive Validierung von Prozess und Werkzeugen, Testen des Produkts
Angabe aller Standorte einschließlich aller an der Entwicklung und Produktion beteiligten Lieferanten und Unterauftragnehmer | X
X | X
– |
| Qualitätssicherung | Anforderungen an Leistungsfähigkeit und Sicherheit
Referenzen auf Nachweise aller grundlegenden Anforderungen Verifizierung und Validierung zum Nachweis der grundlegenden Anforderungen mit Begründung der Wahl insbesondere betreffend Sicherheit Performanz, Genauigkeit (bei Messfunktion), Interoperabilität Nennung aller harmonisierten Normen oder anderer Standards | X
X X X | X
X X X |
| Risikomanagement | Risiko-Nutzenanalyse
Risikomanagementplan Risikoakzeptanz Risikoanalyse (auch durch Gebrauchstauglichkeit, Produktion und nachgelagerte Phase) Maßnahmen zur Risikokontrolle Ergebnisse des Risikomanagements und Maßnahmen | X
X X X X X | X
X X (X) X X |
| Daten aus Forschung und Entwicklung (prä-klinische und klinische Daten) | Ergebnisse von Vortests (z.B. Labor, Simulation, Tierversuche) der Biokompatibilität, elektrischen Sicherheit, biologischen Sicherheit und Software (s. nächster Punkt)
Klinische Bewertung | X
X | (X)
X |
| Software | Verifizierung
Validierung Architektur Entwicklungsprozess Tests mit verschiedener Hardware Tests „inhouse“ und in Gebrauchsumgebung | X
X X X X X | (X)
X – X – – |
| Postproduktion | Hier nicht näher beleuchtet | X | X |
4. Änderungen bei der Klassifizierung und „Zulassung“
Es gibt viele Änderung bei den Konformitätsbewertungsverfahren: Ein Verfahren vergleichbar Anhang VI der MDD gibt es nicht mehr. Die gute Nachricht ist, dass es die Konformitätsbewertung durch die Hersteller überhaupt noch gibt, also eine Zulassung ohne eine europäische Medizinprodukte-Behörde, wie man es von Arzneimitteln kennt.
Neu sind die Scrutiny-Verfahren: Benannte Stellen können verpflichtet werden, jeden neuen Antrag auf Konformitätsbewertung für ein Produkt mit hohem Risiko an eine Expertenkommission (die Medical Device Coordination Group, MDCG) zu melden.
Die Klassifizierung einiger Produkte ändert sich. So müssen eine Reihe von Implantaten, die bisher in Klasse IIb eingestuft waren, nun die Anforderungen von Klasse-III-Produkten erfüllen. Software fällt kaum noch in die Klasse I. Lesen Sie zur Regel 11 hier einen ausführlichen Artikel.
Es wird eine EU-weite Vereinheitlichung der Tätigkeit und der Prüfbescheinigungen der Benannten Stelle geben (MDR-Zertifikat).
5. Sonstige Unterschiede zwischen MDR und MDD
Die Aufbewahrungsdauer der Dokumentation hat die MDR von 5 auf 10 Jahre verdoppelt.
Die MDR führt mit der „für die Einhaltung der Regulierungsvorschriften verantwortliche Person“ eine neue Rolle ein. Sowohl die Hersteller als auch deren Bevollmächtigte müssen über solche eine Person responsible for Regulatory Compliance verfügen.
Die Datenbank EUDAMED wird erheblich ausgeweitet und – nachdem sie bisher staatlichen Institutionen vorbehalten war – nun teilweise auch Herstellern, Benannten Stellen sowie der Öffentlichkeit zugänglich gemacht.
Die Benannten Stellen machen zudem klar, dass die bisherigen PLM/OEM-Konstruktionen nicht mehr akzeptiert werden. Lesen Siehier mehr zu OEM und hier zu Tipps zum Umgang mit den neuen Anforderungen.
Die MDR enthält spezifische Anforderungen an Händler und Importeure.
Die MDR stellt andere Anforderungen an die Konformitätserklärung. Erfahren Sie im weiteren Beitrag dazu, wie Sie mit Produkten umgehen, für die die Konformität noch nach den EU-Richtlinien erklärt wurde.
Lesen Sie in einem separaten Beitrag mehr über die neuen und geänderten Anforderungen und Auswirkungen der MDR auf die Entwicklung medizinischer Software.
Das Johner Institut unterstützt Medizinproduktehersteller auch von „Legacy Devices“, schnell und ohne unnötigen Aufwände die Anforderungen der MDR zu erfüllen.
Interesse? Dann nehmen Sie gleich Kontakt auf.


