
Die Begriffe Wartung, Instandhaltung, Instandsetzung, Inspektion, Service und sicherheitstechnische Kontrolle sind nicht synonym. Aber alle bezeichnen Aktivitäten im Lebenszyklus von Medizinprodukten, die dem Ziel dienen, die Sicherheit, Leistungsfähigkeit und Wirksamkeit dieser Produkte auch nach der Inverkehrbringung zu gewährleisten.
Hersteller und Betreiber müssen die regulatorischen Anforderungen an die Wartung bzw. Instandhaltung erfüllen. Diese Vorgaben sind oft vage. Geben Normen wie die ISO 5137 und die DIN EN 62353 nützliche Hilfestellungen?
1. Die Begriffe: Instandhaltung, Wartung, Instandsetzung
a) Definitionen
Weder die ISO 13485 noch die MDR definieren Begriffe wie Instandhaltung und Wartung. Sie verwenden sie aber.
Die MPBetreibV gibt Hinweise, wie die Begriffe zu verstehen sind:
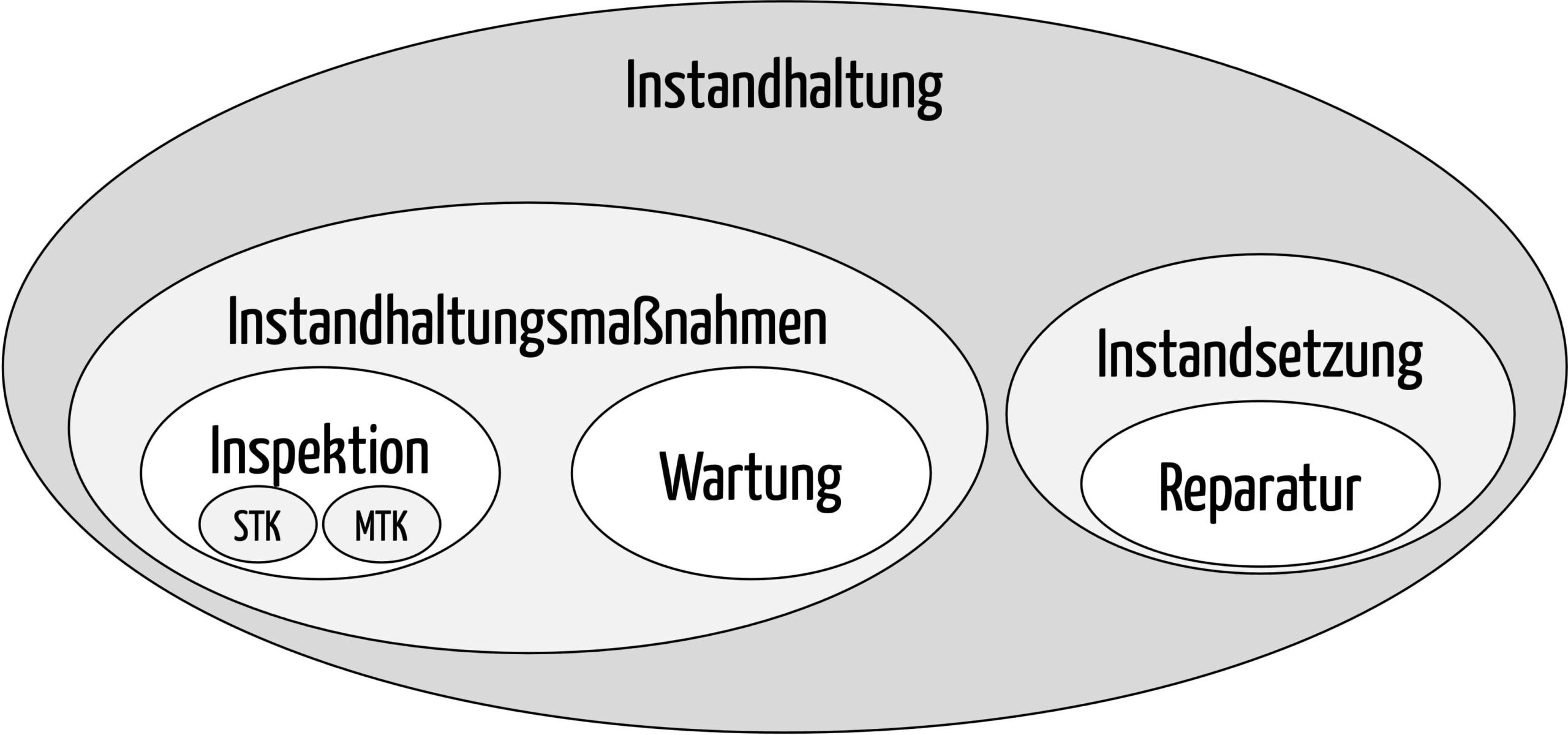
Die Instandhaltung von Medizinprodukten umfasst insbesondere Instandhaltungsmaßnahmen und die Instandsetzung. Instandhaltungsmaßnahmen sind insbesondere Inspektionen und Wartungen, die erforderlich sind, um den sicheren und ordnungsgemäßen Betrieb der Medizinprodukte fortwährend zu gewährleisten.
Quelle § 7 MPBetreibV
Die Instandhaltung ist somit der Oberbegriff, der die Wartung umfasst.
Auch die IEC folgt dieser Hierarchie:
combination of all technical and management actions intended to retain an item in, or restore it to, a state in which it can perform as required
Quelle: IEC Elektropedia
Diese Instandhaltungsmaßnahmen umfassen die Wartung:
vorbeugende Instandhaltung
Quelle: IEC Electropedia
Maßnahmen zur Verzögerung des Abbaus des vorhandenen Abnutzungsvorrates
Quelle: DIN 31051:2003
Auch die Inspektion zählt zu den Instandhaltungsmaßnahmen.
Bestimmung der Konformität mit festgelegten Anforderungen
Quelle: ISO 9000:2015 3.11.7
b) Beispiele für die Wartung / Instandhaltung von Medizingeräten
Wenn eine Medizintechnikerin an einem Gerät misst, ob es noch die versprochene Leistung erbringt, wäre das eine Inspektion. Als Inspektion zählen auch Überprüfungen, ob
- Verbrauchsartikel wie Schmiermittel ersetzt werden müssen,
- Verschleißartikel ausgetauscht werden müssen,
- lose Kabel wieder befestigt und Schrauben wieder angezogen werden müssen und
- das Gerät über das vorgesehene Maß hinaus verschmutzt ist und gereinigt werden muss.
Die von der MPBetreibV geforderten messtechnischen und sicherheitstechnischen Kontrollen sind Sonderfälle dieser Inspektionen (s. Abb. 1).
Der Austausch von Teilen, die drohen, bald abgenutzt zu sein oder kaputtzugehen, wäre eine Wartungsmaßnahme. Beispiele für solche Teile sind Schläuche, Gummimuffen und Dichtungen.
Falls defekte oder abgenutzte Teile ersetzt oder repariert (z. B. geklebt) werden, spricht man von Instandsetzung.
2. Regulatorische Anforderungen an die
Instandhaltung / Wartung
a) MDR (betrifft v. a. die Hersteller)
Die MDR stellt keine präzisen Forderungen an die Instandhaltung bzw. Wartung. Unter anderem fordert sie, dass „die Produkte so ausgelegt und hergestellt werden, dass […] Risiken ausgeschlossen oder so weit wie möglich reduziert werden“. Dazu zählen explizit:
Risiken aufgrund der Alterung der verwendeten Werkstoffe oder der nachlassenden Genauigkeit einer Mess- oder Kontrolleinrichtung, die sich dadurch ergeben, dass keine Wartung oder Kalibrierung vorgenommen werden kann (z. B. bei Implantaten)
MDR Anhang I, Abschnitt 14.2.
Wenn eine Instandhaltung durchgeführt werden soll, fordert die Verordnung:
Die Produkte werden so ausgelegt und hergestellt, dass Einstellung, Kalibrierung und Instandhaltung sicher und wirksam durchgeführt werden können.
MDR Anhang I, Abschnitt 14.4
Bei strahlenden Produkten müssen die Hersteller in der Betriebsanleitung die „Wartungsverfahren“ beschreiben (MDR Anhang I, Abschnitt 16.1).
Auch die Gebrauchsanweisung soll „gegebenenfalls“ die „Art und Häufigkeit präventiver und regelmäßiger Instandhaltungsmaßnahmen“ beschreiben (MDR Anhang I, Abschnitt 23.4.k)).
b) MPBetreibV (betrifft die Betreiber)
Die MPBetreibV verlangt von Betreibern wie Krankenhäusern:
- „Instandhaltungsmaßnahmen sind unter Berücksichtigung der Angaben des Herstellers durchzuführen.“
- Die Kompetenz der daran beteiligten Personen
- Die Dokumentation dieser Tätigkeiten (im Medizinproduktebuch)
- Sicherheitstechnische Kontrollen (STK) für Produkte der Anlage I
- Messtechnische Kontrollen (MTK) für Produkte der Anlage II
Die MPBetreibV verlangt nicht nur die STK und MTK für die jeweiligen Produktgruppen, sondern explizit auch die Durchführung von Instandhaltungsmaßnahmen nach Vorgaben der Hersteller.
Auch Betreiber in anderen Ländern als Deutschland müssen den Vorgaben der Hersteller folgen. Diese Vorgaben dienen nämlich als risikominimierende Maßnahmen.
Lesen Sie hier mehr zur MPBetreibV und den messtechnischen und sicherheitstechnischen Kontrollen.
c) ISO 13485 (betrifft v. a. die Hersteller)
Die ISO 13485 verpflichtet die Medizinproduktehersteller nicht generell zur Instandhaltung ihrer Produkte. Falls die Hersteller allerdings solch eine Instandhaltung vorsehen, müssen sie Folgendes gewährleisten:
- Beschreibung der Verfahren zur Instandhaltung
- Ggf. dafür notwendige Referenzmaterialien und Referenzmessungen
- Eine Lenkung der Lieferanten, die diese Tätigkeiten im Auftrag des Herstellers durchführen
- Die Dokumentation der Tätigkeiten
d) IEC 62304 und IEC 82304 (betrifft v.a. die Hersteller)
Die IEC 62304 fordert im Kapitel 6 einen Wartungsprozess, ähnliches tut die IEC 82304 im Kapitel 8.2. Allerdings ist der Begriff Wartung ist im Software-Kontext anders zu verstehen. Denn die Software-Wartung bedeutet im Gegensatz zu anderen Produkten immer eine Änderungen am „Design“ (dem Entwurf) des Produkts.
Dennoch gibt es einen Bezug dieser Normen zur Wartung/Instandhaltung im Sinne dieses Artikels: Die IEC 82304 verlangt im Kapitel 7.2.3.1, dass der Hersteller in den Begleitmaterialien die Wartungsanforderungen spezifiziert. Dazu zählt die Norm Anforderungen an:
- Umgang mit Auditlogs (bewerten, löschen)
- Pflege der Datenbank
- Wechsel von Speichermedien
e) Weitere Vorgaben für Deutschland
Im Kontext der Instandhaltung und Wartung sollten die Betreiber die folgenden regulatorischen Vorgaben beachten:
- Röntgen- und Strahlenschutzverordnung
- DIN EN 50678 VDE 0701:2021-02 und DIN EN 50699 VDE 0702:2021-06 zur Instandsetzung und Wiederholungsprüfung elektrischer Geräte
- DGUV Vorschrift 3 mit dem Titel „Elektrische Anlagen und Betriebsmittel“
f) FDA (betrifft v. a. die Hersteller)
Die FDA spezifiziert im 21 CFR 820.200 die Anforderungen an den „Service“ noch etwas genauer. So schreibt dieses Gesetz die Inhalte des Serviceberichts vor (z. B. Gerätetyp, Geräteinstanz, Datum, durchführende Person, Tätigkeiten, Ergebnisse).
Allerdings wird die FDA die Anforderungen der ISO 13485 übernehmen.
3. ISO CD 5137
Die Norm lag laut ISO-Webseite zum Zeitpunkt des Schreibens dieses Artikels im Status „Committee Draft“ vor. Ihre Qualität ist/war noch unzureichend, sodass von einem Kauf abgeraten wird. Die Norm kann als Ideenquelle dienen; für die Umsetzung erscheint sie in der aktuellen Form nicht geeignet.
Wenn Sie es eilig haben, überspringen Sie dieses Kapitel.
a) Anwendungsbereich und Struktur
Die ISO 5137 ist eine Norm, die sich speziell an Gesundheitseinrichtungen wie Krankenhäuser und Labore wendet. Sie umfasst 29 Kapitel und 10 Anhänge. Ein Mindmap mit der Kapitelstruktur finden Sie in diesem Dokument:
b) Wesentliche Anforderungen
bESP: biomedial Engineering Services Provider
Die Norm verlangt einen „biomedial Engineering Services Provider“ (bESP), der angestellt ist oder als externer Dienstleister agieren kann.
any in-house or third-party personnel, organization, responsible to carry out, implement or manage medical equipment maintenance activities
Im Kapitel 5 („Responsibilities“) bestimmt die ISO 5137 die Aufgaben dieser „biomedical Engineering Services Providers“ (bESP). Diese reichen vom Implementieren des Wartungsprogramms über die Durchführung jährlicher interner Audits bis zur Sicherstellung notwendiger Ressourcen.
Die Norm legt keine konkreten Anforderungen an die Kompetenz der bESPs fest. Sie bestimmt aber im Kapitel 6 die Kriterien, anhand derer die Gesundheitseinrichtung diese Kompetenzen bestimmen muss.
Übersicht über die Kapitel
Die Tabelle im oben verlinkten Dokument
- verschafft exemplarisch eine Übersicht über die Anforderungen der ersten 20 Kapitel und
- kommentiert diese.
c) Bewertung der ISO CD 5127
Es bleibt zu hoffen, dass die Norm in dieser Form nicht verabschiedet wird. Zu umfangreich sind die Verbesserungspotenziale:
- Die Norm verwechselt Ziele und Anforderungen.
- Die Anforderungen wirken willkürlich zusammengestellt. Sie sind teilweise banal, teilweise auf viele Produktklassen nicht anwendbar.
- Ein konsistenter, prozessorientierter Ansatz, gar Vorgaben zur Integration in ein QM-System, über das die meisten Gesundheitseinrichtungen verfügen müssen, ist nicht erkennbar.
- Die Struktur der Norm ist mit 30 Kapiteln unübersichtlich bis inkonsistent.
- Die Norm definiert Begriffe und Abkürzungen zu spät, falsch oder gar nicht. Sie verwendet Begriffe inkonsistent, sogar im Zusammenhang mit den von ihr selbst normativ referenzierten Normen.
- Die Norm widerspricht mit ihren Konzepten (z. B. den Tags) international gültigen Vorgaben.
Damit kann die Norm in der aktuellen Form als Quelle für Anregungen dienen, eignet sich aber noch nicht für eine Umsetzung in der Praxis.
4. DIN EN 62353 / VDE 0751-1
Die DIN EN 62353/ VDE 0751-1 trägt den Titel „Wiederholungsprüfungen und Prüfung nach Instandsetzung von medizinischen elektrischen Geräten“.
a) Anwendungsbereich
Diese Norm ist wohltuend prägnant und klar strukturiert (s. Abb. 2). Sie wendet sich jedoch nur an medizinisch elektrische Geräte oder medizinisch elektrische Systeme – d. h. an die Produkte bzw. Systeme, die in den Anwendungsbereich der IEC 60601-1 fallen.
Bei anderen Produkten wie Laborprodukten kann die Norm aber auch angewendet werden.
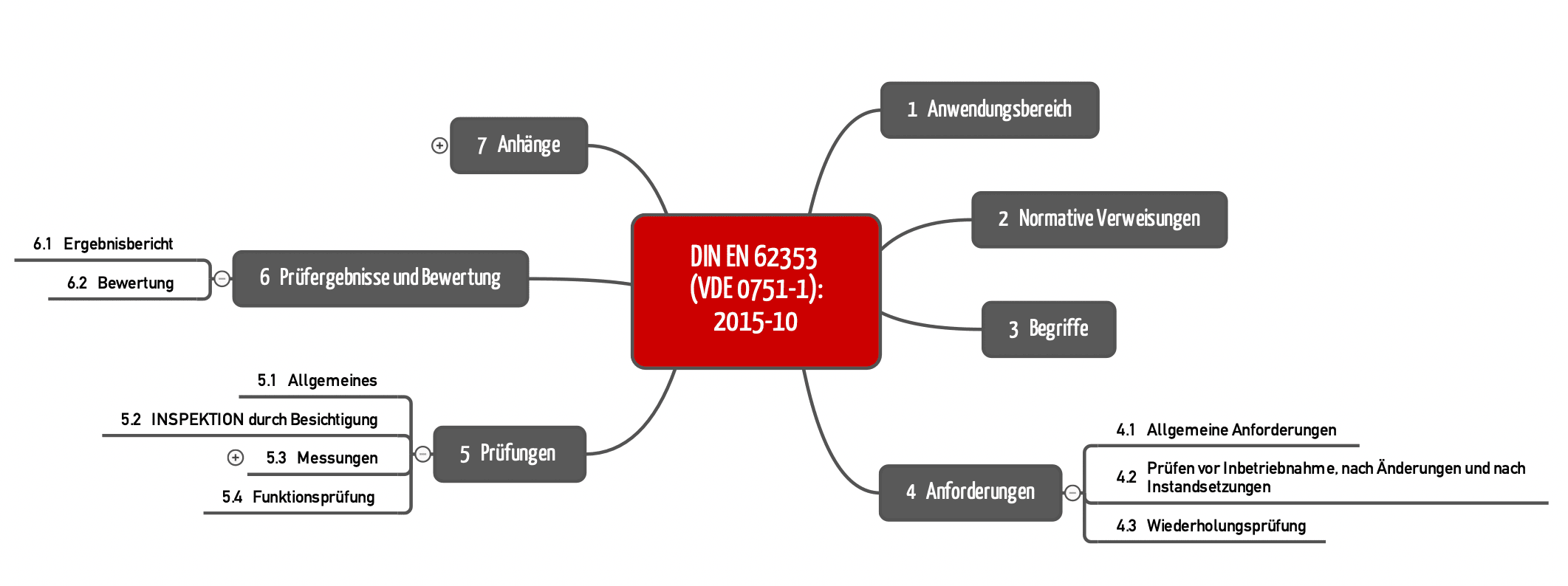
b) Anforderungen
Die Norm stellt „nur“ Anforderungen an die Prüfung dieser Produkte. Sie gibt keine Vorgaben an die Instandsetzung, z. B. an die Reparatur, an den Austausch oder an die Modifikation von Komponenten. Allerdings soll sie für Prüfungen nach (!) einer Reparatur genutzt werden.
Die DIN EN 62353 legt fest, wie die Prüfungen (Messungen, Inspektionen, Funktionsprüfungen) durchgeführt werden sollen.
Beim Schwedischen Institut für Normen können Sie kostenlos einen umfangreichen Preview einer älteren Version der Norm einsehen.
Für wenige Euro können Sie die IEC 62353 hier beziehen.
5. Tipps und Checklisten
a) Tipps für Hersteller
Überlegungen zur Entwicklungsphase
Hersteller sollten beim Festlegen der Vorgaben für die Wartung bzw. Instandhaltung folgende Einflussfaktoren berücksichtigen (s. Abb. 3):
- Die Zweckbestimmung und die Stakeholder-Anforderungen (z. B. Lebensdauer, Nutzungskontext, Nutzungshäufigkeit und die vorgesehenen Nutzer) bestimmen die Produktanforderungen. Die Lebensdauer entspricht sogar einer Produktanforderung.
- Die Produktanforderungen bestimmen den Entwurf des Produkts. Lebensdauer und Nutzungshäufigkeit müssen die Hersteller beispielsweise bei der Auswahl von Materialien und Komponenten berücksichtigen.
- Wenn das Produkt entworfen ist, wird klar, welche Maßnahmen notwendig sind, um die Risiken während der Nutzungsphase weiterhin zu beherrschen, hier: welche Instandhaltungstätigkeiten notwendig sind.
- Am besten sind inhärent sichere Produkte. Das sind Produkte, die auch inhärent keine Risiken durch die Wartung haben, weil z. B. keine Wartung notwendig ist.
- Die spezifizierten Instandhaltungstätigkeiten sind wiederum Teil des bestimmungsgemäßen Gebrauchs, der entsprechend formuliert sein muss.
- Das Risikomanagement beeinflusst alle Tätigkeiten. Es hilft insbesondere, die notwendigen Instandhaltungsmaßnahmen zu bestimmen.
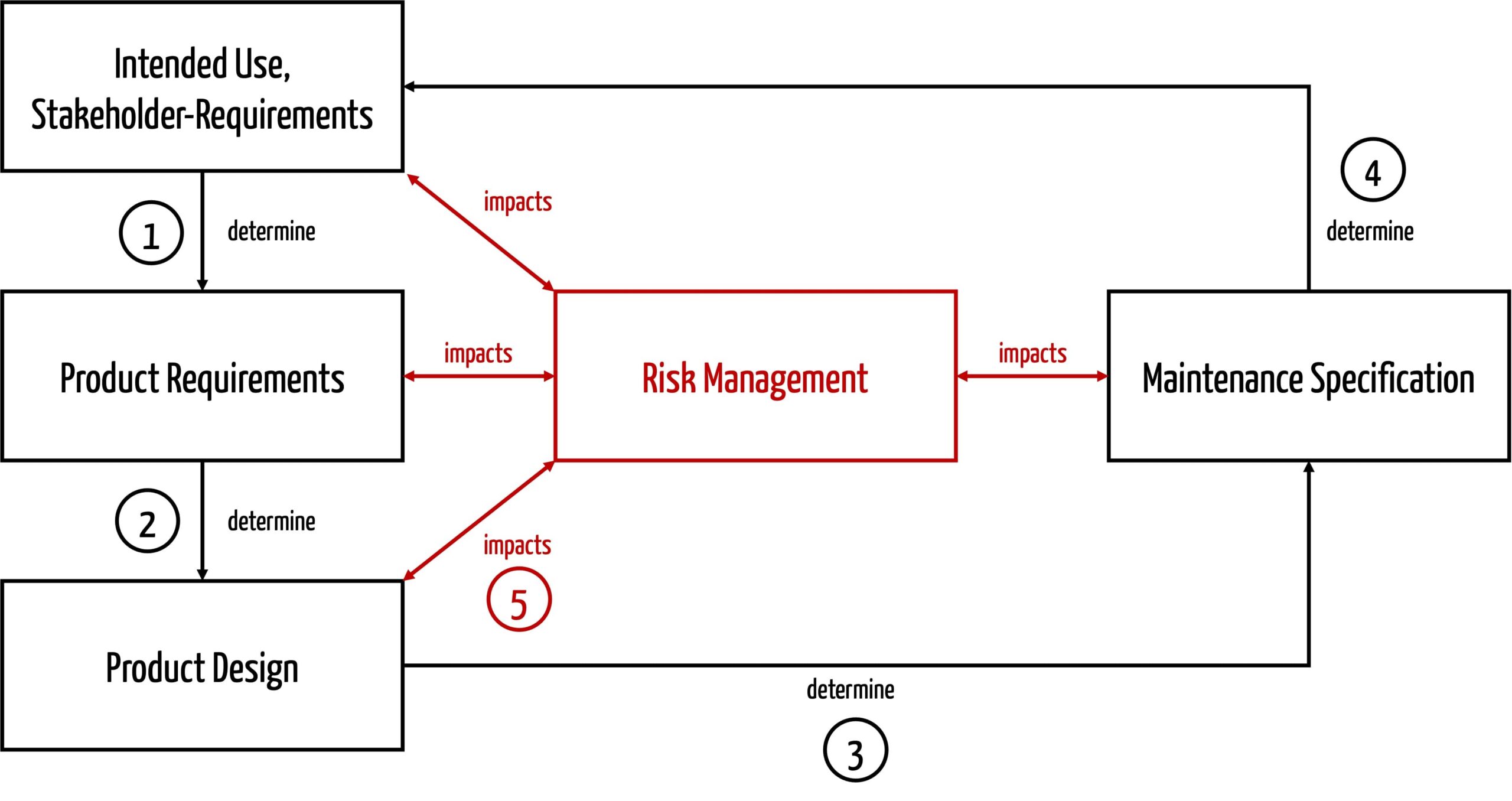
Beim Festlegen der Instandhaltungsmaßnahmen sollten die Hersteller auf Folgendes achten:
- Die Instandhaltungsmaßnahmen müssen aus dem Risikomanagement abgeleitet werden.
- Die möglichen Maßnahmen sind nicht auf messtechnische und sicherheitstechnische Kontrollen beschränkt.
- Hersteller müssen die Gebrauchstauglichkeit der Instandhaltungsanweisungen (konform IEC 62366-1) gewährleisten. Dazu müssen sie die Kompetenz der Personen, die mit diesen Anweisungen arbeiten, kennen, festlegen und ggf. schaffen.
- Die Forderungen der ISO 13485, die beispielsweise mit der Instandhaltung beauftragte „Lieferanten“ betreffen, müssen beachtet werden.
Lesen Sie hier mehr zu den Anforderungen an Begleitmaterialien wie Gebrauchsanweisungen.
Checkliste für die Post-Market-Phase
Hersteller müssen systematisch Informationen sammeln, um Sicherheit, Leistungsfähigkeit und Wirksamkeit ihrer Produkte über den ganzen Produktlebenszyklus zu gewährleisten. Dabei ist zu beachten:
- Wird das Produkt tatsächlich wie vermutet verwendet? (Nutzer, Nutzungshäufigkeit, Nutzungsumgebung)
- Entsprechen die Verschmutzung und der Verschleiß den Annahmen?
- Werden die Instandhaltungstätigkeiten wie spezifiziert durchgeführt? (Frequenz, Kompetenzen, Materialien, konform mit Arbeitsanweisungen)
Checkliste für die Inhalte von Instandhaltungsanweisungen
Die Anweisungen müssen vollständig sein. Typische Elemente einer solchen Instandhaltungsanweisung sind:
- Klare Identifikation des Medizinprodukts, auf das sich die Anweisung bezieht
- Kompetenz der Personen, die für die Instandhaltung vorausgesetzt wird
- Frequenz, Zeitpunkte oder Anlässe der Instandhaltungsmaßnahmen
- Die einzelnen Arbeitsschritte bei der Instandhaltung, ggf. mit Fotos
- Prüfungen (Anweisung, erwartete Ergebnisse mit Pass-fail-Kriterien), um den Erfolg der Instandhaltungsmaßnahmen zu bewerten
- Die Materialien, Bauteile und Werkzeuge, die für die Instandhaltung verwendet werden müssen (ggf. mit Bezugsquelle und Identifikation, z. B. Bestellnummer)
- Vorgaben für die Dokumentation, ggf. mit Formblättern oder Software-Anwendungen
- Meldepflichten an den Hersteller und die Behörden

Profitieren Sie von fundiertem Wissen, das Ihnen dabei hilft, Ihre Arbeit effektiver und sicherer zu gestalten
Unsere innovative E-Learning-Plattform vermittelt Ihnen umfassendes Wissen über die Instandhaltung von Medizinprodukten. Sie lernen Beispiele kennen, welche möglichen Fehlerquellen es gibt und welche Risiken damit verbunden sind. Darüber hinaus zeigen wir Ihnen bewährte Maßnahmen auf, um diese Risiken zu minimieren und zu vermeiden.
b) Tipps für Betreiber
Checkliste für den Einkauf von Produkten
Der Einkauf sollte u. a. auf die folgenden Punkte achten:
- Beschreibt der Hersteller die Instandhaltungsmaßnahmen so präzise, dass das eigene Team sie durchführen kann?
- Erlaubt der Hersteller, dass der Betreiber selbst oder eine von ihm beauftragte Organisation die Instandhaltung durchführt?
- Verfügt das eigene Team über die vorgeschriebenen Kompetenzen?
- Gewährleistet der Hersteller die Versorgung mit Ersatzteilen und Verbrauchsmaterialien sowie die Instandhaltung über während der kompletten Lebensdauer des Produkts?
- Sind die Preise für diese Teile und Materialien sowie für die Instandhaltungsmaßnahmen durch den Hersteller realistisch und bleiben diese während der Lebensdauer des Produkts gewährleistet?
Dass Hersteller auch mit dem „Service“ Geld verdienen wollen, ist legitim. Der Betreiber sollte aber Transparenz über Aufwände und Kosten erlangen können, um eine fundierte Auswahl von Herstellern und Produkten treffen zu können.
Checkliste für die Instandhaltung
Betreiber sollten auch diese Tipps berücksichtigen:
- Die Instandhaltung ist als Prozess im eigenen Qualitätsmanagementsystem integriert. Ein eigenes Qualitätssicherungsprogramm, wie von der ISO 5137 vorgesehen, erzeugt unnötigen Overhead.
- Die vom Hersteller vorgesehenen Instandhaltungsmaßnahmen werden für jedes Produkt ausgeführt, auch wenn es sich nicht um mess- und sicherheitstechnische Kontrollen handelt. Es gibt dafür eine Arbeitsanweisung, die definiert, wer das wann, wie und wie oft durchführt.
- Eine Arbeitsanweisung legt fest, wie die Instandhaltungsmaßnahmen im Medizinproduktebuch dokumentiert werden und wer dies wie und wie häufig prüft.
- Es ist für jedes Produkt dokumentiert, wer die Instandhaltung durchführen darf (Hersteller, Betreiber oder eine von ihm beauftragte Organisation).
- Es ist für jedes Produkt dokumentiert, wer welche Probleme dem Hersteller bzw. dem BfArM meldet.
Mit der Instandhaltung wird leider auch Schindluder von „Wartungs-STK-Organisationen“ getrieben, die den Krankenhäusern und Ärzten überteuerte Wartung/STK verkaufen, mit dem Hinweis, dass dies gesetzlich vorgeschrieben sei, obwohl das im Einzelfall nicht stimmt.
Die Betreiber sind nicht nur auf die Unterstützung durch ihr medizintechnisches Personal angewiesen, sondern auch auf die Unterstützung der Anwender. Dabei ist eine Kultur fatal, in der es akzeptabel ist, abgenutzte Bohrer, Geräte mit brüchigen Netzkabeln und 20 Jahre alte Röntgengeräte zu verwenden.
Das BfArM hat eine umfangreiche FAQ zur MPBetreibV veröffentlicht.
6. FAQ zur Wartung / Instandhaltung von Medizingeräten
a) Wer darf medizinische Geräte prüfen?
Es ist die Aufgabe der Hersteller die Anforderungen an die Wartung / Instandhaltung von Medizingeräten festzulegen. Dazu zählen auch die Anforderungen an die (Kompetenzen der) Personen, welche diese Tätigkeiten durchführen.
Falls die Begleitmaterialien zum Medizinprodukt erklären, wie die Wartung bzw. Instandhaltung durchgeführt werden muss, kann die Gesundheitseinrichtung davon ausgehen, dass sie diese Tätigkeiten von ausgebildeten Fachkräften wie Medizintechnikern selbst erledigen darf. Fehlen diese Anleitungen jedoch, sollte die Gesundheitseinrichtung beim Hersteller nachfragen.
b) Wie oft müssen medizinische Geräte überprüft werden?
Es gibt zwei Quellen, welche diese Frequenz bestimmen:
- Die Vorgaben des Herstellers, typischerweise in den Begleitmaterialien wie Gebrauchsanweisungen
- Die Medizinprodukte-Betreiberverordnung (MPBetreibV)
Enthalten beide Quellen Angaben zum konkreten Gerät, ist die höhere Frequenz maßgeblich. Falls nur eine Quelle die Frequenz spezifiziert, ist diese zu nutzen. Falls es keine Angaben bzw. Vorgaben gibt, bleibt es dem Betreiber überlassen. Meist erfolgt dann die Instandsetzung anlassbezogen.
c) Was ist die Validierung medizinischer Geräte
Der Begriff Validierung sollte im Kontext von Wartung und Instandhaltung nicht verwendet werden. Die Validierung ist nicht(!) die Prüfung, ob sich ein Medizingerät spezifikationsgemäß verhält. Das wäre eine Verifizierung. Die Validierung ist die Prüfung, ob das medizinische Gerät seine vom Hersteller zugewiesene Zweckbestimmung erfüllt. Ein zentraler Aspekt der Validierung ist die klinische Bewertung.
Beachten Sie weiterführende Artikel zur Verifizierung und Validierung von Medizinprodukten, zur Validierung von IVD und zur Validierung der Gebrauchstauglichkeit.
7. Fazit und Zusammenfassung
Hersteller haben eine hohe Verantwortung für die Sicherheit, Leistungsfähigkeit und Wirksamkeit ihrer Medizinprodukte. Diese Verantwortung betrifft den kompletten Produktlebenszyklus, einschließlich Betrieb und Instandhaltung.
Die Verantwortung liegt jedoch nicht allein bei den Herstellern, sondern auch bei den Betreibern und Anwendern. Gesetzliche Vorgaben wie die MPBetreibV und Normen geben dafür einen Rahmen und Handlungsleitung.
Leider lassen diese Normen ebenso wie die Umgangssprache teilweise klare Konzepte vermissen. Das führt zu Nachlässigkeiten und Unsicherheiten. Daraus ergeben sich einerseits Nachteile für die Patienten, andererseits Nachteile für Betreiber, die sich zu unnötigen und teuren Instandhaltungsmaßnahmen „überreden“ lassen.
Die Instandhaltung muss sowohl bei den Herstellern als auch bei den Betreibern ein risikobasierter Prozess als Teil eines Qualitätsmanagementsystems sein.
Das Johner Institut unterstützt Medizinproduktehersteller dabei, präzise, verständliche sowie normen- und gesetzeskonforme Wartungsanweisungen zu erstellen und auf Gebrauchstauglichkeit zu prüfen. Melden Sie sich gerne über unser Kontaktformular.
Änderungshistorie
- 2024-12-15: Kapitel 6. (FAQ) eingefügt
- 2023-03-05: Kapitel 2.c) zur IEC 62304 und IEC 82304 sowie Hinweis ergänzt, das ISO 5137 im nächsten Entwurfsstatus ist.



Auf https://www.iso.org steht die ISO/CD 5137 im Status „deleted“ ,
heißt wohl, dass die Norm nicht weiter verfolgt wird?
Lieder gibt es auf der Webside keine weiteren Informationen dazu.
Danke für den Hinweis, Herr Balling!
Der „Committee Draft“ scheint damit zurückgezogen zu sein. Seit Ende Dezember 2022 scheint die Norm laut anderer ISO-Webseite aber im nächsten Status „Preparatory“ zu sein.
Diesen Hinweis ergänze ich im Artikel.
Beste Grüße, Christian Johner
Vielen Dank für den informativen Artikel!
Sollten der Vollständigkeit halber hier nicht auch die IEC 62304 sowie die IEC 82304 erwähnt werden? Diese beiden Normen fordern vom Hersteller ebenfalls bestimmte Dinge zur Wartung.
Die IEC 62304 verlangt in Kapitel 6
„Der HERSTELLER muss einen Plan (oder Pläne) für die Software-Wartung festlegen, um die AKTIVITÄTEN und
AUFGABEN des Wartungs-PROZESSES durchzuführen.“
Es muss also für das Produkt einen Wartungsplan geben und es muss ein Wartungsprozess definiert sein.
Die IEC 82304 (welche gemäss unserem Auditor für Medizinprodukte ebenfalls gilt und zwingend zu berücksichtigen ist), erwähnt in Kapitel 8.2 die Softwarewartung (eher unspezifisch). Sie verlangt auch, dass in der technischen Beschreibung (7.2.3.1) die Wartungsanforderungen in der Gebrauchsanweisung angegeben sind.
Freundliche Grüsse
Aurel Baier
Danke Herr Baier für Ihre wertvollen Kommentar!
Sie haben absolut Recht, dass die IEC 82304 auch Anforderungen stellt. Das ergänze ich.
Nochmals vielen Dank!
Viele Grüße, Christian Johner
Nur ein ganz kleiner Hinweis: Ein CD ist kein „Common Draft“ sondern ein „Committee Draft“.
Sie haben absolut Recht, lieber Herr Müller!
Den Fehler konnte ich Dank Ihrer Hilfe sofort korrigieren!
Besten Dank und herzliche Grüße, Christian Johner
Liebes Herr Prof. Johner,
ich habe eine Frage zur Instandsetzung wieder verwendbarer Medizinprodukte, die eine UDI tragen, genauer gesagt zum Anhang VI, Teil C, Ziffer 4.10 der MDR. Demnach muss der UDI-Träger „dauerhaft angebracht und nach jedem Verfahren, das zur Vorbereitung des Produkts für die nachfolgende Verwendung durchgeführt wird, während der gesamten erwarteten Lebensdauer des Produkts lesbar sein.“
Es wird in der Praxis beobachtet, dass der UDI-Code oftmals so stark abgenutzt oder gar beschädigt ist, dass er nicht lesbar und somit auch nicht einlesbar ist.
Wie ist mit der Reparatur von solchen Medizinprodukten umzugehen?
1. Kann eine Instandsetzung erfolgen und der UDI-Code weggelassen werden, da es Pflicht des Herstellers bzw. Betreibers ist, die Aufrechterhaltung des Codes sicherzustellen?
2. Eine Instandsetzung kann in diesen Fällen nicht erfolgen (obwohl technisch möglich), da der UDI-Code der Medizinprodukte nach der Reparatur nicht dauerhaft lesbar ist.
Vorab vielen Dank für Ihre Antwort,
M. Mayer
Unter der Überschrift im Kapitel „3c) MPBetreibV (betrifft die Betreiber)“ verstehe ich, dass deren Paragraphen den Hersteller nicht betreffen. In §7 Abs (2) und Abs (4) der MPBetreibV steht sinngemäss, dass Personen, Betriebe und Einrichtungen, welche eine Instandhaltung resp. Prüfung als Beauftragte durchführen, die Anforderungen von §5 erfüllen müssen.
Das interpretiere ich so, dass wenn der Hersteller eine Instandhaltung resp. Prüfung im Auftrag eines Betreibers durchführt, §5 erfüllen muss und dann zumindest dieser Paragraph auch auf den Hersteller anwendbar ist.
Stimmt da meine Interpretation oder ist der Hersteller komplett ausserhalb der Anwenbarkeit der MPBetreibV ?
Sehr geehrter Herr Meschberger,
ich sehe das genau wie Sie: Ein Hersteller, der als Dienstleister im Auftrag eines Betreibers agiert, sollte sicherstellen, dass der Betreiber seinen regulatorischen Anforderungen gerecht wird. D.h. es greifen für diese Fälle (indirekt) die regulatorischen Anforderungen an die Betreiber, nicht an die Hersteller. Dass dieser Dienstleister zufällig auch der Hersteller ist, ändert daran nichts.
Viele Grüße
Christian Johner
Liebes Herr Prof. Johner,
ich habe eine Frage zur Instandsetzung wieder verwendbarer Medizinprodukte, die eine UDI tragen, genauer gesagt zum Anhang VI, Teil C, Ziffer 4.10 der MDR. Demnach muss der UDI-Träger „dauerhaft angebracht und nach jedem Verfahren, das zur Vorbereitung des Produkts für die nachfolgende Verwendung durchgeführt wird, während der gesamten erwarteten Lebensdauer des Produkts lesbar sein.“
Es wird in der Praxis beobachtet, dass der UDI-Code oftmals so stark abgenutzt oder gar beschädigt ist, dass er nicht lesbar und somit auch nicht einlesbar ist.
Wie ist mit der Reparatur von solchen Medizinprodukten umzugehen?
1. Kann eine Instandsetzung erfolgen und der UDI-Code weggelassen werden, da es Pflicht des Herstellers bzw. Betreibers ist, die Aufrechterhaltung des Codes sicherzustellen?
2. Eine Instandsetzung kann in diesen Fällen nicht erfolgen (obwohl technisch möglich), da der UDI-Code der Medizinprodukte nach der Reparatur nicht dauerhaft lesbar ist.
Vorab vielen Dank für Ihre Antwort,
M. Mayer
Liebe Frau Mayer,
danke für die Frage!
Um die Frage zu beantworten, müsste ich die Rolle der Person kennen, die aufbereitet bzw. repariert. Falls das ein externer Dienstleister im Auftrag eines Krankenhauses ist, dann bleibt es in der Verantwortung des Krankenhauses, die Konformität aufrechtzuerhalten.
Das heißt aber nicht, das beispielsweise Hersteller keine Pflichten hätten. Sie sind im Rahmen der Post-Market-Surveillance verpflichtet, genau solche Probleme zu identifizieren und zu beheben. Und auch das Krankenhaus (der Betreiber) muss Probleme melden.
Wenn ich ein „Reperaturdienstleister“ wäre, würde ich das Produkt reparieren und nachweisbar den Betreiber informieren, dass das Medizinprodukt die gesetzlichen Anforderungen (hier ans Labeling) nicht mehr erfüllt. Im Rahmen Ihrer Prozesses bzw. Ihres QM-Systems haben Sie die Anforderungen an die Traceability sicherzustellen. Das ist ohne eine UDI ggf. schwierig.
Viele Grüße
Christian Johner
Sehr geehrter Prof. Johner,
im Zuge der Beschäftigung mit den einschlägigen Paragraphen der MDR für Wirtschaftsaktuere und einigen Beobachtungen in unsrem MDR Audit stellt sich mir die Frage welche Verpflichtungen bezüglich der Überwachung von Installation und Wartungsmassnahmen der Hersteller hat bezüglich Geräten die über Distributoren in Drittländer vertrieben werden.
Beispiel: eines unserer Geräte (nach EU Recht CE zertifiziert) wird von einem Händler erworben um es in einem nicht-EU Land weiter zu vertreiben (die erforderliche Registrierung/zertifizierung wurde veranlasst und liegt vor).
Der Händler (Distributor) hat eine QSV bezüglich der Anforderungen der MDR mit uns, dort ist aber nichts bezüglich Wartung etc. erwähnt.
Der Distributor erhält alle Informationen zur Installation und Wartung ebenfalls laut Vereinbarung von uns.
Sind wir als Hersteller dann dazu verpflichtet die Reports zur ordnungsgemäßen Installation und Wartung beim Endkundent proaktiv einzusammeln bzw. regelmäßig einsenden zu lassen und vorzuhalten?
Dies kann bei weltweitem Vertrieb un einigen Ländern recht aufwändig werden. Auch haben wir (also unsere RA) nicht den Überblick über die unzähligen lokalen Entsprechungen der MBetreibV.
Oder reicht es wenn wir den Distributor dazu verpflichten, Installation und Wartung nach seinen lokalen Gesetzgebungen durchzuführen und entsprechende Aufzeichnungen vorzuhalten im Falle zB eines Recalls?
Also vergleichbar des Handlings mit Tickets/Complaints.
Aus der MDR oder dem Blue Guide kann ich daraus nichts ableiten. Vielleicht können Sie uns da weiterhelfen.
Viele Grüße
Fritz Vollmer
Sehr geehrter Herr Vollmer,
danke für Ihre wichtige Frage!
Die Antwort auf Ihre Frage hängt von der regulatorischen Brille ab.
Die MDR hat die Sicherheit der europäischen Patienten im Blick. Daher stellt sie keine konkreten Anforderungen an die Wartung bzw. Instandhaltung in anderen Rechtsbereichen. Allerdings verlangt sie, dass Sie
Ich schätze es nicht für ausreichend ein, wenn Sie Aufzeichnungen von Installationsaktivitäten nur fordern im Fall der Fälle. Es wäre hilfreich, wenn Sie Rückmeldungen über Risiken einfordern. Hinweise über den (unerwartet häufigen) Ausfall von Bauteilen oder anderen Problemen können Sie ggf. auch durch das Ersatzteilmanagement nachverfolgen.
Beste Grüße
Christian Johner
Sehr geehrter Herr Johner,
vielen Dank für Ihre Ausführungen, das deckt sich auch mit meinem Verständnis der Anforderungen aus QMS und PMS Sicht und gibt mir eine gute Ausrichtung für die erforderlichen QSV Anpassungen.
Viele Grüße
Fritz Vollmer
Danke für Ihre Rückmeldung, lieber Herr Vollmer!
Mich freut es, dass wir ein gemeinsames Verständnis haben.
Nochmals vielen Dank!
Beste Grüße, Christian Johner
Sehr geehrter Hr. Prof. Johner!
Gibt es Anforderungen / Vorschriften and die Konstrukteure / Entwickler eines neuen Medizinprodukts bezüglich Reparierbarkeit und Rezyklierbarkeit (speziell im Zusammenhang mit dem EU Green Deal)?
Lieber Herr Gassner,
Das ist eine sehr gute Frage. Die EU hat verschiedene Verordnungen im Rahmen des Green Deals veröffentlicht, darunter die neue Batterieverordnung, die auch für Medizinprodukte gilt. Weitere bekannte Verordnungen sind die WEEE und REACH. Es gibt auch Normen, die einige Anforderungen spezifizieren, wie zum Beispiel die IEC 60601-1-9. Eine umfassende Zusammenstellung dieser Verordnungen und Normen kann ich hier leider nicht bieten. Melden Sie sich einfach, wenn ich helfen kann.
Liebe Grüsse, Mario Klessascheck
Sehr geehrter Herr Johner,
wenn ein Medizinprodukt, das eine Sonderanfertigung ist, von einem vom Hersteller autorisierten Maintenance Center gewartet und ggf. repariert wird, bei wem liegt die Verantwortung für das Produkt? Für den Fall, dass nach der Instandhaltung das Produkt den Dienst versagt und der Patient evtl. zu Schaden kommt, bei wem liegt dann die Verantwortung? Wer kann dafür haftbar gemacht werden? Der Hersteller oder das Maintenance Center?
Ein weiterer Punkt : Lebensdauer- Verlängert sich mit der Instandsetzung die Lebensdauer des MP ab diesem Zeitpunkt?
Vielen Dank im Voraus für Ihre Unterstützung.
Mit freundlichen Grüßen
Christine Graß
Sehr geehrte Frau Graß,
die Verantwortung zur Einhaltung der MDR trägt zunächst der Hersteller, neben weiteren Wirtschaftsakteuren (Händler, Importeure, etc.). Wenn das Maintenance Center das Produkt nicht nur überprüft und repariert, sondern als neu aufbereitet, würde dieses auch die Rolle des Herstellers einnehmen (siehe Definition „Hersteller“ im Artikel 2 (30.) MDR). Bezüglich Produkthaftung trifft die MDR keine Regelungen. Entsprechende Regelungen finden sich beispielsweise im Produkthaftungsgesetz. Wir als Johner Institut dürfen Ihnen leider keine Rechtsberatung anbieten. Dazu wenden Sie sich am besten an eine Anwaltskanzlei im Bereich Produkthaftung (z.B. an Dr. Handorn, http://www.produktkanzlei.com).
Zur Lebensdauer: Ob sich die Lebensdauer durch eine Instandsetzung verlängert, muss der Hersteller selbst ermitteln bzw. festlegen. Dazu kann dieser beispielsweise Daten aus der Marktüberwachung nutzen. Bei einer vollständigen Neuaufbereitung (siehe Definition in der MDR) würde eine neue Lebensdauer starten.
Herzliche Grüße
Luca Salvatore
Sehr geehrte Damen und Herren,
ich habe eine Frage bezüglich der Wartung von Klasse 1 MP, für welche einen Zeitraum von 3 Nutzungsjahren vorgesehen ist.
Wo ist definiert, für welche Produkte eine Wartung durchgeführt werden muss? Kann vom Hersteller garantiert werden, dass das jeweilige Produkt für den Nutzungszeitraum von 3 Jahren ohne Wartung genutzt werden kann?
Bitte um eine Rückmeldung zu meinem Anliegen.
Vielen Dank!
Sehr geehrte Frau Stadlmayr,
danke für die spannende Frage!
Die Lebens- bzw. Nutzungsdauer ist eine Angabe, die vom Hersteller stammen muss. Ein Hersteller kann so etwas durchaus garantieren. Das setzt voraus, dass er die Sicherheit und Wirksamkeit seiner Produkte über diesen Zeitraum auch ohne Wartung nachweisen kann. Je nach Produkt müssen dazu Alterungstests durchgeführt werden, welche die drei Jahre Nutzung simulieren.
Es gibt somit keine gesetzliche Vorgabe, die eine Garantie des Herstellers von drei Jahren verbieten würde. Eine Abhängigkeit einer zugesagten Nutzungsdauer von der Klasse gibt es ebenfalls nicht.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner
Danke für den Artikel. In eigen Aspekten ist er aber nicht so präzise, wie ich mir das Klinik interner Instandhalter wünsche. Zum einen ist auch die „Verbesserung“ gemäss DIN 31051eine Massnahme der Instandhaltung. Dieser Sachverhalt ist für uns sehr wichtig, da wir in vielen Fällen „Schwachstellen beseitigen“ können ohne die Konformität der Produkte zu beeinträchtigen.
Ein anderer wichtiger Aspekt ist der der „Wirksamkeit“ der vom Hersteller definierten Instandhaltungsmassnahmen. Es gibt diverse Forschungs- und Erfahrungsberichte darüber, dass die Sicherheit von Medizinprodukten auch zu gewährleisten ist, wenn man (punktuell) von den Hersteller Empfehlungen abweicht. Auch die VDI 5707 weisst darauf hin, dass die Hersteller-Vorgaben vielfach „Maximal-Anforderungen“ darstellen. Gesetze und Normen lassen eine Evidenz basierte Instandhaltung (EBM) von Medizinprodukten zu und in einigen Gesundheitseinrichtungen haben sich solche Strategien etabliert. Gerade in Zeiten von knappen Ressourcen und steigenden Kosten im Gesundheitswesen, sollten wir solchen Instandhaltungs-Strategien aufzeigen.
Sehr geehrter Herr Römmelt,
ich verstehe Ihre Überlegungen sehr gut. Vielen Dank dafür.
Die Anforderungen der Hersteller sind in der Tat oft „Maximal-Anforderungen“. Wenn Sie allerdings davon ableiten, gehen Sie ein haftungsrechtliches Risiko ein. Unabhängig davon, ob Ihre tatsächlich umgesetzten Maßnahmen die Sicherheit der Patienten erhöhen, erniedrigen oder nicht beeinflussen.
Die Definition der DIN 31051 des Begriffs Instandhaltung deckt sich nicht mit dem Verständnis des Medizinprodukterechts. Daher würde ich diese nur behutsam verwenden. Eine Verbesserung bezieht sich in der Regel auf die Sicherheit, Leistungsfähigkeit oder Wirksamkeit von Medizinprodukten. Deren Nachweise obliegen den Herstellern.
Beste Grüße, Christian Johner
Sehr geehrter Herr Dr. Johner,
wer ist für die elektrische Prüfung (EN 62353 / DIN VDE 0571-1) verantwortlich? Der Lieferant/Hersteller oder der Betreiber?
Wir führen aktuell die Wartung/Reparatur der von uns vertriebenen Medizingeräte (nicht Hersteller sondern Distributor) durch und überprüfen nicht automatisch auch die elektrische Sicherheit nach diesen Tätigkeiten.
Muss der Betreiber (Kunde) uns dafür beauftragen oder sind wir in der Pflicht, diese elektrische Prüfung durchzuführen?
Vielen Dank und liebe Grüße
Christian Möller
Lieber Herr Möller,
Generell ist der Betreiber für die Wiederholungsprüfungen verantwortlich. Er kann diese selbst durchführen, oder andere Organisationen (beispielsweise Sie) damit beauftragen. Die elektrischen Prüfungen dürfen nur von Fachpersonen durchgeführt werden. Der Hersteller sollte in seiner Gebrauchsanweisung beschreiben, welche Prüfungen bei einer Wiederholungsprüfung oder nach einer Reparatur durchzuführen sind. Falls nicht, hilft die IEC EN 62353 bei der Durchführung.
beste Grüsse, Mario Klessascheck