
Die Zulassung von Medizinprodukten in Japan bedeutet für europäische Hersteller eine große Hürde. Sie sollten davor aber nicht zurückschrecken, denn Japan gehört zu den 10 größten Märkten der Welt.
Welche Voraussetzungen Sie erfüllen müssen und wie Sie die Hürden am besten überwinden, verrät Ihnen dieser Beitrag.
A) Regulatorischer Rahmen bei der Zulassung
1. Behörden
Die Pharmaceuticals and Medical Devices Agency (PMDA) ist Japans regulatorische Behörde. Sie untersteht dem Ministry of Health, Labour and Welfare (MHLW).
Die Mission des MHLW besteht darin, die Bevölkerung Japans vor Gesundheitsschäden zu bewahren, die durch mangelnde Sicherheit, Wirksamkeit oder Qualität von pharmazeutischen Produkten und Medizinprodukten hervorgerufen werden können. Zu den Aufgaben des MHLW gehören im Rahmen der Medizinprodukteregulierung:
- Durchführung von Hersteller-Registrierungen
- Vergabe von Lizenzen für bestimmte Akteure (MAH, Händler und Reparaturdienstleister)
- Verabschiedung von Verordnungen (Ministerial Ordinances), Leitfäden und Industriestandards
- Finale Freigabe von Medizinprodukte-Zulassungen
- Überwachung der PMDA
Die eigentlichen Aufgaben innerhalb der Zulassungsverfahren von Medikamenten, Medizinprodukten und regenerativen medizinischen Produkten im japanischen Markt übernimmt die 2004 gegründete PMDA. Zu den wichtigsten Aufgaben der PMDA gehören:
- Durchführung der Zulassungsverfahren einschließlich der regulatorischen Prüfung
- Beratung zu klinischen Studien und angestrebten Zulassungen
- Inspektionen von Herstellern zur Einhaltung der Good Manufacturing Practices (GMP)
- Marktüberwachung
2. Regularien
Seit 2014 ist der Pharmaceutical and Medical Device Act (PMD Act) das wichtigste Gesetz. Er löste das „Pharmaceutical Affairs Law“ (PAL) ab.
Der PMD Act beinhaltet Anforderungen an die Inverkehrbringung, die Qualitätssicherung und die Gewährleistung der Wirksamkeit und Sicherheit von Medikamenten, Medizinprodukten, regenerativen medizinischen Produkten, zellulären Therapien, Gentherapien und Kosmetika.
In der Hierarchie der Regularien folgen dem PMD Act zunächst „Cabinet Ordinances“, dann „Ministerial Ordinances/Notification“ und schließlich „Administrative Notices“. Diese Regularien sind für die Zulassung von Medizinprodukten in Japan relevant. Die „Administrative Notices“ sind auf der Ebene der MDCG- und FDA-Guidances anzusiedeln und nicht gesetzlich verbindlich einzuhalten.

B) In 8 Schritten zur Zulassung
1. Schritt: QM-System etablieren
Sie müssen als ausländischer Hersteller bei der Zulassung von Medizinprodukten in Japan kein „Home Country Approval“ nachweisen. Allerdings ist Ihnen ein ISO-13485-Zertifikat sehr hilfreich, um die Anforderung an das japanische Qualitätsmanagement (J-QMS) nachzuweisen.
Die Vorgaben an das Qualitätsmanagementsystem finden sich in der in 2021 revidierten Ministerial Ordinance No. 169 (MO 169). Die MO 169 ist im Wesentlichen mit der ISO 13485:2016 harmonisiert (Vergleichstabelle Kapitel 2). Allerdings befinden sich in Kapitel 3 zusätzliche Anforderungen, die Sie als Hersteller sowie Ihr Bevollmächtigter (MAH) zu erfüllen haben. Diese beinhalten Anforderungen an Aufbewahrungsfristen, Vigilanz und Regelungen zur Kommunikation zwischen Hersteller und Bevollmächtigtem.
2. Schritt: Notwendige Rollen etablieren
Marketing Authorisation Holder (MAH)
Als nächstes sind Sie als Hersteller verpflichtet, einen Bevollmächtigten vor Ort zu benennen, den sogenannten Marketing Authorisation Holder (MAH). Der MAH übernimmt die Haftung für Ihre Produkte in Japan und ist der Eigner der Zulassungen.
Ihr MAH muss nicht nur einen Sitz in Japan haben und Ihre Zulassungsunterlagen einreichen; er hat zusätzlich die Gesamtverantwortung für das QM-System und ist nach der Zulassung Ihrer Produkte auch für die Produkt- bzw. Chargenfreigabe, die Post-Market Surveillance und die Vigilanz verantwortlich.
Nicht jeder Bürger darf die Tätigkeit eines MAH ausüben. Der MAH muss zuerst eine Lizenz (Business License namens KYOKA) beim MHLW beantragen. Es gibt folgende Typen von Lizenzen:
- First Class: Der MAH darf für sämtliche Medizinprodukte (Klassen I, II, III, IV) benannt werden (zur Klassifizierung lesen Sie unten mehr).
- Second Class: Der MAH darf für Medizinprodukte der Klassen I und II benannt werden.
- Third Class: Der MAH darf nur für Medizinprodukte der Klasse I benannt werden.
Ein MAH muss mindestens die folgenden drei Rollen ausfüllen:
- General Marketing Supervisor/Director
- Quality Manager
- Safety Manager
Bei einem Third Class-MAH darf eine Person alle drei Rollen einnehmen. Bei einem Second Class-MAH darf der General Marketing Supervisor zusätzlich die Rolle des Quality Managers oder des Safety Managers übernehmen. Bei einem First Class-MAH darf der General Marketing Supervisor nicht der Safety Manager sein. Somit bedarf es bei First und Second Class-MAH mindestens zwei Personen.
Die Rolle des MAH könnte z.B. von Ihrem Distributor, einer Niederlassung vor Ort oder einem unabhängigen Unternehmen übernommen werden.
Designated Marketing Authorisation Holder (D-MAH)
Alternativ zum MAH können Sie einen Designated Marketing Authorisation Holder (D-MAH) benennen. Unterschied zum MAH ist dabei, dass Sie in dieser Konstellation als Hersteller selbst der Zertifikatseigner sind. Der D-MAH übernimmt ausschließlich die Verantwortung für das QMS einschließlich Produkt- bzw. Chargenfreigabe, Post-Market Surveillance und Vigilanz. Somit sind Ihr Zertifikat und die Produktzulassung nicht betroffen, wenn Sie den D-MAH wechseln möchten.
Dieses Vorgehen mit dem D-MAH bietet sich erfahrungsgemäß für kleinere und mittlere Unternehmen an. Hingegen bietet es sich für größere Unternehmen an, den japanischen Sitz bzw. die Tochterfirma als MAH zu wählen. Bei Klasse I-Produkten ist die D-MAH-Konstellation leider nicht möglich.
Weitere Rollen
Schließlich sind Sie als Hersteller bereits bei der Produktzulassung verpflichtet, den Domestic Warehouse Manufacturer zu benennen. Dieser ist für die Lagerung und den Versand in Japan verantwortlich
Anschließend müssen Sie vor dem Import festlegen, wer für Distribution und Reparatur verantwortlich ist.
Auch für diese Rollen (Transport, Vertrieb, Reparatur) bedarf es einer Registrierung über das MHLW.
3. Schritt: Sich als Hersteller registrieren
Vor der Zulassung Ihres Medizinprodukts in Japan müssen Sie sich mithilfe des Marketing Authorisation Holders (MAH) beim Ministry of Health, Labour and Welfare (MHLW) als Foreign Manufacturer registrieren. Dabei sind alle Herstellungsstätten zu registrieren, die für die Entwicklung und finale Montage bzw. Fertigung verantwortlich sind. Komponenten-Hersteller müssen sich nicht registrieren.
Die Beantragung zur Registrierung wird durch Formular 63-5 begleitet. Für die Registrierung sind u.a. die folgenden Informationen notwendig:
- Allgemeine Information (Name und Adresse des Herstellers)
- Liste aller Produkte
- Selbsterklärung durch Ihren Manager
- Information über die Verantwortlichen Personen
- Information über die Produktionsstätte
Die Registrierung ist ein eher einfacher administrativer Prozess. Die Dauer von der Einreichung bis zur Registrierung beträgt etwa 30 Tage. Die Registrierung ist für fünf Jahre gültig und muss danach erneuert werden. Änderungen von z.B. Adresse oder Kontaktperson sind innerhalb von 30 Tagen zu melden.
4. Schritt: Klassifizierung des Medizinprodukts vornehmen
Der PMD Act unterscheidet drei Typen von Medizinprodukten: General, Controlled und Specially-Controlled Medical Device. Diese werden in vier Klassen unterteilt: I (geringes Risiko), II, III, IV (hohes Risiko).
Die folgende Tabelle gibt Beispiele für die Klassifizierung von Medizinprodukten in Japan.
| Klasse | Beschreibung | Beispiel |
|---|---|---|
| I: General Medical Device | Niedriges Risiko | Skalpell, Pinzette |
| II: Controlled Medical Device | Relativ niedriges Risiko | Endoskopie, dentale Legierungsmittel, Ultraschallgerät, Magnetresonanztomograph |
| III: Specially-Controlled Medical Device | Hohes Risiko | Dialysator, künstliches Knochentransplantat |
| IV: Specially-Controlled Medical Device | Invasiv, potenziell lebensbedrohlich | Stent, Herzschrittmacher, künstliche Herzklappe |
Im Gegensatz zur EU erfolgt die Klassifizierung ähnlich wie in den USA über vorgegebene Produkt-Codes: Die Japanese Medical Device Nomenclature (JMDN) Die JMDN wurde im Jahr 2005 veröffentlicht und basiert auf der vierten Version der Global Medical Device Nomenclature (GMDN).
Die JMDN enthält nicht nur den achtstelligen JMDN-Code selbst, sondern auch
- den generellen Namen des Produkttyps,
- Definitionen,
- die risikobasierte Klassifizierung gemäß GHTF,
- Informationen, ob die QMS-Anforderungen einzuhalten sind,
- die Klassifizierungsregeln und
- anwendbare Zulassungs- bzw. Review-Kriterien (z.B. anzuwendende Normen).
Falls kein geeigneter JMDN-Code vorhanden ist, muss risikobasiert anhand der GHTF-Regeln klassifiziert werden.
5. Schritt: Zulassungsverfahren auswählen
Neben der Klassifizierung gibt es weitere Einflussfaktoren auf die Wahl des Zulassungsverfahrens.
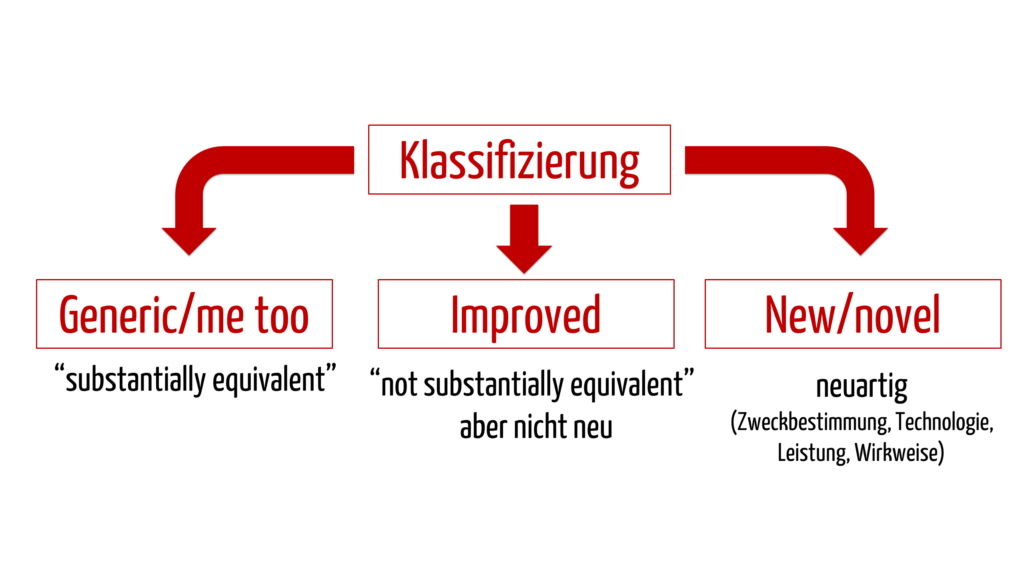
- Vergleichsprodukte in Japan: Es wird unterschieden zwischen generic/me too, improved und novel. Generic bedeutet, dass es bereits identische oder im Wesentlichen ähnliche Vergleichsprodukte auf dem japanischen Markt gibt („substantially equivalent“ zu einem „predicate device“). Improved sind Medizinprodukte, für welche keine geeigneten predicate devices vorhanden sind. Bei novel handelt es sich um neuartige Medizinprodukte, welche sich wesentlich unterscheiden bezüglich Zweckbestimmung, Technologie, Wirkweise oder Leistung.
- Zertifizierungsstandard: Für eine Vielzahl an JMDN-Codes sind Zertifizierungsstandards angegeben. In diesem Fall darf die Zulassung über ein Third-Party Review durch eine unabhängige Zertifizierungsstelle (siehe weiter unten) erfolgen. Zum Beispiel wäre für JMDN-Code „36208000 – Mobile ultrasound imaging system“ der anwendbare Zertifizierungsstandard JIS_T_0601-2-37, welcher der IEC 60601-2-37 entspricht.
Basierend auf der Klassifizierung sowie den beiden genannten Einflussfaktoren wählen Sie anschließend eines der drei Zulassungsverfahren.
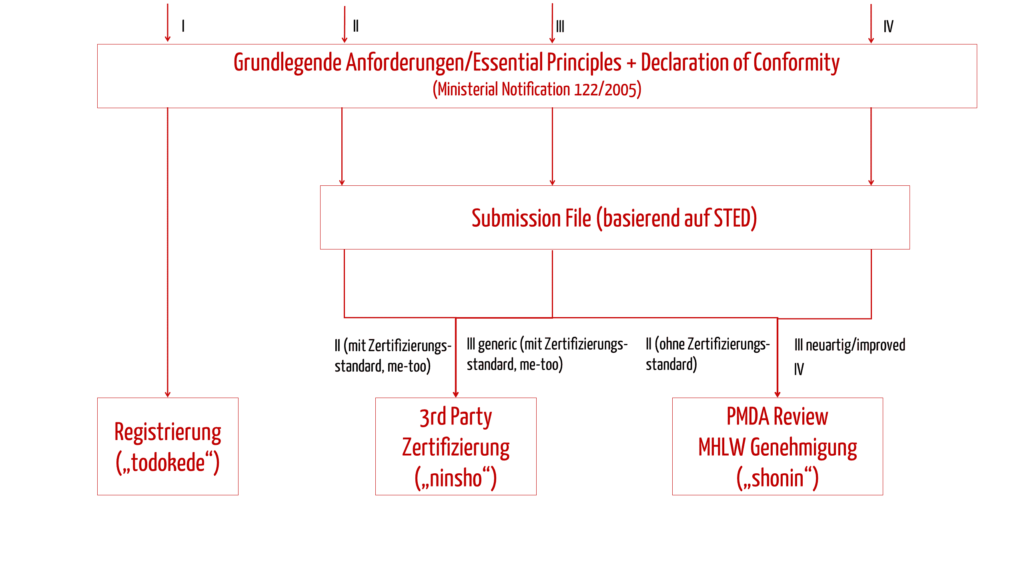
| Name des Verfahrens | Anwendbarkeit | Kurzbeschreibung des Verfahrens |
|---|---|---|
| TODOKEDE | Klasse I | Einfache Registrierung der Produkte mit Konformitätserklärung (ähnlich wie bei Klasse I in der EU) |
| NINSHO | Klasse II und III generic, mit Zertifizierungsstandards | Ein RCB (Registered Certification Body) übernimmt als „Third Party“ die Prüfung der Dokumente der Zulassung und der Konformität mit den grundlegenden Anforderungen („Essential Principles“). |
| SHONIN | Klasse II und III ohne Klassifizierungsstandards sowie Klasse IV | PMDA prüft die Dokumente der Zulassung und die Konformität mit den „Essential Principles“. |
Todokede
Der MAH meldet im Namen des Herstellers das Klasse-I-Produkt bei der PMDA. Dazu reicht der MAH neben Angaben zum Hersteller (Herstellungsstätten, Herstellungsverfahren, Registrierungsnummer), einer allgemeinen Beschreibung des Medizinprodukts (u.a. Zweckbestimmung, Handelsname, Wirkweise, Leistungsmerkmale, Spezifikationen, Anforderungen an die Lagerung und Haltbarkeit) auch die Gebrauchsanweisung auf Japanisch ein. Zusätzlich müssen Sie als Hersteller immer die grundlegenden Anforderungen bzw. die Essential Principles einhalten. Eine Prüfung der Einhaltung findet im Todokede-Verfahren allerdings nicht statt. Die Registrierung ist typischerweise innerhalb eines Monats erledigt.
Ninsho
Beim Ninsho-Verfahren, auch bekannt als „pre-market certification“, sind sogenannte Registered Certification Bodies (RCB) für die Dokumentprüfung verantwortlich. RCBs sind japanische „Benannte Stellen“, die den Zulassungsprozess in Form eines Third-Party-Reviews übernehmen.
Der RCB prüft dazu die Zulassungsdokumente auf Einhaltung der Zertifizierungsstandards und der grundlegenden Anforderungen (Essential Principles) und erteilt im positiven Fall die Zertifizierung. Zu den RCBs zählen der TÜV Süd, TÜV Rheinland, BSI Group, SGS und DEKRA (vollständige Liste auf Japanisch).
Das Ninsho-Verfahren ist für die meisten Klasse II-Produkte und für einige Klasse III-Produkte anwendbar. Durchschnittlich dauert die Prüfung der Zulassungsdokumente durch RCBs (für Klasse-II- und Klasse-III-Produkte) vier Monate.
Shonin
Falls Sie Medizinprodukte der Klassen II und III ohne Zertifizierungsstandards oder Produkte der Klasse IV in Japan zulassen wollen, müssen Sie das strenge Shonin-Verfahren, auch bekannt als „pre-market approval“ (PMA), mit Prüfung und Zulassung über die PMDA durchlaufen. Für die meisten Standalone-Software-Produkte (SaMD) sind aktuell keine Zertifizierungsstandards vorhanden, wodurch ein Shonin-Verfahren anzuwenden ist.
Für generic/me-too-Produkte sind im Regelfall sogenannte Approval Standards oder Approval Guidelines definiert. Diese sind in der JMDN-Datenbank den einzelnen JMDN-Codes zugeordnet. Sie enthalten beispielsweise anzuwendende Normen oder produktspezifische Guidances.
Die Dauer des Shonin-Verfahrens beträgt etwa 12 Monate ohne Einberechnung der Vorbereitungszeit der Einreichungsunterlagen oder der Durchführung von klinischen Studien.
Die offiziellen Kosten für die japanische Zulassung können Sie dieser Liste (nur auf Japanisch) entnehmen.
Beachten Sie, dass auch Produkte der Klassen I und II, für die noch kein JMDN-Code festgelegt wurde, dieses PMA-Verfahren durchlaufen müssen.
6. Schritt: Zulassungsunterlagen zusammenstellen und einreichen
Klinische Bewertung erstellen (kann klinische Studie bedingen)
Vor der Zulassung Ihres Medizinprodukts in Japan sind Sie ebenso wie in Europa verpflichtet, im Rahmen einer klinischen Bewertung den Nachweis des klinischen Nutzens, der Leistungsfähigkeit und der Sicherheit Ihres Produkts zu erbringen.
Japan erlaubt es ebenso wie Europa, diesen Nachweis unter Umständen anhand von Literaturdaten zu erbringen. Für generic/me-too-Produkte sind keine klinischen Nachweise einzureichen. Hingegen sind für neuartige Medizinprodukte klinische Studien notwendig. Für improved-Produkte, deren Sicherheit und Leistung nicht durch präklinische Daten und Literaturdaten ausreichend nachgewiesen wird, sind ebenfalls Daten aus klinischen Studien einzureichen.
Es ist nicht in allen Fällen zwingend, mögliche klinische Studien auch in Japan durchzuführen bzw. klinischen Daten aus Japan zu verwenden.
Für die Anerkennung von ausländischen klinischen Studien ist es ratsam, dass sich Ihr MAH vorab mit der Behörde austauscht.
Zulassungsunterlagen allgemein
Alle Dokumente, die Sie bei der Zulassung einreichen, müssen auf Japanisch vorliegen.
Die Einreichungsunterlagen basieren auf dem international anerkannten STED-Format des IMDRF. Zusätzlich zur zusammenfassenden STED-Dokumentation werden folgende Anhänge verlangt:
A – Entwicklungshistorie (frühere Produktversionen, Zulassungen weltweit) B – Produktspezifikationen
B – Produktspezifikationen
C – Daten zur Stabilität und Haltbarkeit
D – Einhaltung der anzuwendenden Normen und grundlegenden Anforderungen
E – Daten zu Performance-Tests
F – Risikoanalyse
G – Herstellung (Prozess, Überwachung, Sterilisation)
H – Klinische Daten
Es gibt in der Praxis eine offene Kommunikation mit der Behörde, sobald dort mit der Prüfung der Dokumente begonnen wurde. Das heißt, dass die vorgesehene Prüfdauer der Dokumente bei Rückfragen der Behörde nicht unterbrochen wird.
Auch eine Zulassung der klinischen Prüfung erfolgt durch diese interaktive Kommunikation relativ schnell.
Als Hersteller tun Sie gut daran, Ihre Marketingstrategie für Japan frühzeitig zu planen. Das ermöglicht es Ihnen, bereits während der Zulassung Ihr Produkt auf Messen zu präsentieren.
Sie unterschreiben für diesen Zweck einen Vertrag mit dem Distributor, verkaufen das Produkt jedoch erst nach erfolgter Zulassung.
7. Schritt: QMS-Inspektion beantragen
Vor Produktfreigabe wird die PMDA oder der RCB (abhängig vom Zulassungsverfahren) das QM-System des MAH und der registrierten Herstellungsstätten, d.h. auch jenes des ausländischen Herstellers, auditieren. Für jede Produktfamilie wird dabei ein eigenständiges Audit durchgeführt. Die Beantragung erfolgt durch den MAH. Die Überprüfung erfolgt entweder in Form einer Vor-Ort-Inspektion oder in Form eines Dokumenten-Audits. Für welches Verfahren sich die Behörde entscheidet, ist u.a. abhängig von einer existierenden Zertifizierung nach ISO 13485, von den Ergebnissen der letzten Audits, von gemeldeten Vorkommnissen und Rückrufen sowie der Komplexität des Herstellungsprozesses. Bei Software-Herstellern erfolgt im Regelfall ein Dokumenten-Audit.
Beim Dokumenten-Audit prüft die Behörde folgende Unterlagen:
- Übersicht Herstellungsstätte
- Organigramm
- QM-Handbuch
- Liste der QM-Dokumente
- Herstellungsprozess inkl. Validierung
- MAH-Vertrag und Vigilanz-Verfahren
- Falls angefragt: Zusammenfassung der Medizinprodukteakte
Seit 2015 nimmt Japan am Medical Device Single Audit Program (MDSAP) teil. Seit Ende der Pilotphase akzeptiert Japan die MDSAP-Audit-Berichte. Somit sind im Regelfall keine weiteren Dokumente einzureichen. Hersteller, die sich nach MDSAP auditieren lassen, ersparen sich somit redundante Audits.
8. Schritt: Post-Market-Phase planen
Änderung von Medizinprodukten nach der Zulassung
Genau wie in vielen anderen Ländern müssen Sie als Hersteller eine „Change Notification“ einreichen, wenn sich Ihre Produkte oder weitere Angaben ändern. In Japan wird dabei unterschieden nach partial change und minor change. Partial changes beinhalten Änderungen an der Zweckbestimmung oder sonstige Änderungen mit Einfluss auf die Sicherheit oder Wirksamkeit des Produkts, z.B. Änderungen am Funktionsprinzip, an Materialien oder am User Interface. Minor changes decken alle Änderungen ab, die nicht unter die Definition eines partial change fallen. Für minor changes reicht eine einfache Meldung innerhalb von 30 Tagen nach Durchführung der Änderung. Für partial changes ist eine „partial change application“, also eine erneute Einreichung notwendig.
Post-Market-Surveillance
Die Pflicht zur Post-Market-Surveillance betrifft auch Ihren MAH. Diesen machen die japanischen Regularien dafür verantwortlich, Vorkommnisse mit Medizinprodukten an den jeweiligen Hersteller und die Behörde weiterzuleiten. Die Vorgaben finden sich in der „Good Vigilance Practice“ (Ministerial Ordinance 135).
Als Medizinproduktehersteller sind Sie verpflichtet, Ihren MAH bei der Durchführung der Marktüberwachung zu unterstützen. Entsprechende Regelungen im Vertrag mit Ihrem MAH oder im Rahmen Ihres QM-Systems sind dringend zu empfehlen.
Einen guten Überblick über die Anforderungen an das Melden von Vorkommnissen und an die Rückrufe erhält man in Kapitel 4 des MDSAP Companion Document. Dieses referenziert die relevanten regulatorischen Anforderungen. Generell müssen Sie Ihren MAH unverzüglich über schwerwiegende Vorkommnisse (auch im Ausland) informieren.
C) Unterstützung bei der Zulassung
Das Johner Institut unterstützt Medizinproduktehersteller bei der Zulassung ihrer Medizinprodukte in Japan in folgenden Phasen:
- Produkte klassifizieren
- Zulassungsverfahren auswählen
- Zulassungsunterlagen erstellen und prüfen
- MAH finden
Melden Sie sich z.B. über dieses Kontaktformular bei uns, wenn Sie mit Ihren Medizinprodukten im japanischen Markt schnell erfolgreich sein möchten.
D) Fazit, Zusammenfassung
Die Hürden für die Zulassung von Medizinprodukten in Japan sind nicht mehr so unüberwindbar wie früher. Dazu trägt auch das MDSAP-Verfahren bei. Zudem gibt es viele Parallelen zur Zulassung in Europa und den USA.
Allerdings bedeuten die Rollen wie der MAH, ggf. der DMAH und der DSM sowie die Pflicht, die Unterlagen auf Japanisch einzureichen, nennenswerte Aufwände.
Dafür ist der japanische Markt groß und stabil. Laut BVMED liegt Japan bereits auf dem achten Platz der Länder, in die deutsche Medizinproduktehersteller exportieren.



Sehr geehrter Herr Johner,
wir sind Hersteller eines sterilen IVDs, welches wir auch in Japan verkaufen. Sowohl wir als Hersteller als auch unser Sterilisationsunternehmen haben ein gültiges Registration certificate of foreign medical device manufacturer.
Ein OEM Kunde von uns, für den wir auch ein steriles IVD herstellen will sein Produkt jetzt auch in Japan registrieren. Das OEM Produkt wird beim gleichen Sterilisationsunternehmen sterilisiert. Kann sich der OEM Hersteller auf das bestehende Registration Certificate vom Sterilisationsunternehmen beziehen, oder muss es sich nochmal registrieren? Es haben nur wir einen Vertrag mit dem Sterilisationsunternehmen und nicht unser OEM Kunde.
Hat die Klassifizierung des IVDs einen Einfluss darauf, welche Dokumente für eine Foreign Medical Device Manufacturer Registrierung bereitgestellt werden müssen? Bei unserer Registrierung (Klasse II Produkt) mussten wir weniger Dokumente bereitstellen als unser OEM Kunde (Klasse III Produkt).
Vielen Dank für Ihre Hilfe!
Mit freundlichen Grüßen,
Martina
Guten Tag,
die Registrierung wird immer vom (D-)MAH des jeweiligen Herstellers beantragt. Somit wäre dafür der (D-)MAH des OEMS verantwortlich. Die Registrierung des Sterilisationsunternehmens sollte in diesem Fall anerkannt werden, so dass sich dieses nicht erneut registrieren muss. Generell hat die Klassifizierung keinen Einfluss darauf, welche Dokumente bei der Herstellerregistrierung eingereicht werden müssen.
Herzliche Grüße
Luca Salvatore
Sehr geehrter Herr Johner,
gibt es in Japan ein Special Access Programm, wie z.B. in Australien und Kanada, damit ein Angehöriger eines Gesundheitsberufes ein Medizinprodukt beantragen kann, welches in Japan nicht zum Verkauf steht?
Vielen Danke und Grüße
Lena
Hallo Lena,
laut unserem Experten vor Ort in Japan gibt es dort ein ähnliches Programm:
„Doctors and dentists can import unapproved medical devices for their own use with their own patients. My understanding is that they need to get approval and that the criteria is that there are no alternatives available in Japan.
The doctor or dentist takes full responsibility for the device.“
Viele Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen Dank für diese sehr hilfreiche Übersicht zur Zulassung in Japan.
Meine Recherche ergab, dass die Japanische Standards Association Group die internationalen Normen prinzipiell anerkennt. Dennoch scheint sich die JSA die Möglichkeit offen zu lassen, Normen als „not equivalent“ zu kennzeichnen. Wie kann man prüfen, ob eine internationale Norm auch in Japan gilt? Ein konkretes Beispiel wäre die IEC 62353.
Vielen herzlichen Dank im Voraus und freundliche Grüße
Peter Schenk
Sehr geehrter Herr Schenk,
eventuell hilft Ihnen ein Blick auf die Webseite der JSA: https://webdesk.jsa.or.jp/books/W11M0090/index
Dort können Sie nach Normen suchen (international und JST) mit Äquivalenzvermerk. Wenn Sie dort beispielsweise nach ISO 14971 suchen, dann bekommen Sie als Ergebnis die Referenz zur japanischen Version (JIS T 14971:2020) und den Hinweis zur Äquivalenz. In diesem Fall IDT, was für „identisch“ steht. Die IEC 62353 ist nicht als japanische Norm verfügbar. Generell empfehle ich, die anwendbaren Normen für ein konkretes Produkt über die JMDN-Datenbank zu recherchieren über folgenden Link: https://www.std.pmda.go.jp/stdDB/index_en.html
Dort bekommen Sie, abhängig vom Produkttyp, anwendbare „certification/approval“ Kriterien angezeigt. Zu diesen Kriterien gehören vor allem Normen. Bei einigen Produkten liefert Ihnen die Datenbank sogar eine vorausgefüllte Essential Requirements Checkliste mit Angabe der anzuwendenden Normen, z.B. https://www.std.pmda.go.jp/stdDB/Data_en/MDStd/CerStd/1300487_04_2019_en.pdf (Nuclear medicine system workstation).
Beste Grüße
Luca Salvatore
Sehr geehrter Herr Salvatore,
vielen herzlichen Dank für die hilfreichen Informationen.
Wenn für ein Produkt in der JMDN-Datenbank keine spezifischen „certification/approval“ Kriterien genannt werden, nehme ich an, dass es diese auch nicht gibt und entsprechend selbst definiert, bzw. passende gewählt werden müssen?!
Freundliche Grüße
Peter Schenk
Sehr geehrter Herr Schenk,
ganz genau. Falls es diese nicht gibt, müssen vom Hersteller Kriterien definiert werden zum Nachweis der Sicherheit und Wirksamkeit.
Beste Grüße
Luca Salvatore
Hallo, wir haben eine Registrierung unserer Produkte in Japan erfolgreich durchgeführt. Leider sind die Zertifikate nur auf Japanisch. Wissen Sie wie die Anforderungen hinsichtlich der Dokumentenablage bei dem deutschen Hersteller ist? Muss es eine beglaubigte Übersetzung einer Behörde sein?
Sehr geehrte Frau Kuhlmann,
bezüglich der Registrierung in Japan gibt es von deutscher Seite (Behörden) keine Verpflichtungen für Sie, d.h. z.B. das genannte Zertifikat beglaubigt übersetzen zu lassen. Dennoch sollten Sie als Hersteller den Inhalt des Zertifikats prüfen, wodurch eine Übersetzung sicherlich sinnvoll ist.
Freundliche Grüße
Luca Salvatore
Guten Tag,
was ich nicht ganz verstehe, ist wie die PAL Klassifizierung mit der GHTF Klassifizierung zusammenhängt. Der JMDN Code beinhaltet die Risikoklasse I bis IV und nicht A bis D (GHTF). Kann man sagen, dass I=A, II=B,…usw. ist?
Guten Tag Frau Coßmann,
Ihre Vermutung ist korrekt. Das Mapping finden Sie auf der PMDA-Webseite unter folgenden Link: https://www.std.pmda.go.jp/scripts/stdDB_en/refetc/stdDB_refetc_sum_absbttm.cgi?absdisp=1
Herzliche Grüße
Luca Salvatore
Ist es richtig, dass in Japan neben einer bestehenden Registrierung mit einem MAH auch eine „secondary registration“ oder „dual registration“ mit einem DMAH für das gleiche Produkt möglich ist?
Guten Tag Frau Schulz,
dasselbe Produkt unter demselben License-Holder zu registrieren wird schwer möglich sein, außer man nutzt eventuell verschiedene Produkt- oder Markennamen. Wenn ich es richtig verstehe, wären es in Ihrem Fall aber verschiedene License-Holder (der MAH und Sie als Hersteller). Ich würde dennoch empfehlen, dies mit dem jeweiligen DMAH zu klären bzw. offen zu kommunizieren.
Herzliche Grüße
Luca Salvatore
Guten Tag,
in welcher Verordung wird nun der Aufbau der TD konkret bescheiben (vergleichbar mit ANNEX 2 der MDR)
Viele Grüße
Max Schwanau
Guten Tag Herr Schwanau,
die Anforderungen an die Einreichungsdokumentation findet sich zum einen in der Verordnung zur Umsetzung des PMD Acts (https://www.japaneselawtranslation.go.jp/en/laws/view/3215/en#je_ch3sc1at21, u.a. Artikel 114-19). Detaillierte Informationen findet sich in weiteren Notifications wie 0120 Nr. 9 (https://www.pmda.go.jp/files/000197935.pdf) oder der Review-Checkliste (https://www.pmda.go.jp/files/000267257.pdf). Diese Dokumente sind allerdings nur auf Japanisch verfügbar.
Freundliche Grüße
Luca Salvatore
Habe gerade ein Handbuch auf der PMDA Seite gefunden. Dort sind die Anforderungen der TD zusammengefasst.
https://www.pmda.go.jp/files/000153030.pdf
Lieber Herr Schwanau,
herzlichen Dank für das Teilen dieses hilfreichen Links!
Beste Grüße,
Margret Seidenfaden
Guten Tag,
ich habe eine Frage bezüglich „Zulassungsunterlagen allgemein“. Woher nehmen Sie diese Information, bzw. wo wird dies näher beschrieben?
Viele Grüße
Lieber Herr Merck,
die PMDA hat die Anforderungen an die einzureichende Dokumentation in Abhängigkeit vom Zulassungsverfahren in verschiedenen Notifications veröffentlicht. Diese sind auf der PMDA-Website einsehbar, liegen jedoch nur auf Japanisch vor (z.B: https://www.mhlw.go.jp/file/06-Seisakujouhou-11120000-Iyakushokuhinkyoku/261120kyoku11205.pdf).
Es gibt auch eine Review Checklist, sowie die Informationen des PMD Acts (https://www.japaneselawtranslation.go.jp/en/laws/view/3215/en#je_ch3sc1at21, u.a. Artikel 114-19).
Herzliche Grüße,
Margret Seidenfaden