
ASCA steht für Accreditation Scheme for Conformity Assessment. Das Verfahren soll Konformitätsbewertungen und damit Zulassungsverfahren beschleunigen. Es ist aber weder bei allen Produkten noch bei allen Märkten anwendbar.
Wer von ASCA profitiert und wie das Verfahren abläuft, erläutert dieser Artikel.
1. ASCA – Die Grundlagen
a) Wen betrifft ASCA?
ASCA betrifft v. a. diese Akteure:
- Medizinproduktehersteller, deren Produkte unter die Normenfamilien IEC 60601, IEC 80601, IEC 61010 und ISO 10993 fallen
- Prüfhäuser, die die Produkte auf Konformität mit diesen Normen prüfen
- Akkreditierer (Accreditation Bodies) der Prüfhäuser
- Zulassungsbehörden, insbesondere die FDA
Derzeit ist das Verfahren beschränkt auf die folgenden Zulassungsverfahren in den USA: 510(k), De Novo, Premarket Approval (PMA) und Investigational Device Exemption (IDE).
b) Was ist ASCA?
ASCA ist ein freiwilliges Pilotprogramm für ein neues Akkreditierungssystem, bei dem
- Prüflabore eine zusätzliche Qualifizierung/Akkreditierung durchlaufen und
- Prüfberichte standardisiert werden.
Die FDA vertraut den Prüfmethoden und Ergebnissen (v. a. dem ASCA Summary Test Report) der ASCA-akkreditierten Prüfhäuser und Prüflabore. Sie beabsichtigt nicht, zusätzliche Informationen anzufordern; das beschleunigt die Zulassung.
Der Ablauf des Verfahrens wird weiter unten beschrieben.
c) Warum wird ASCA gebraucht?
Die FDA bemängelt das Auftreten von zu vielen Zwischenfällen mit Medizinprodukten. Als einen Grund dafür hat sie die nicht hinreichende Qualität der Prüfhäuser identifiziert.
Zudem hat die Behörde erkannt, dass Medizinprodukte immer komplexer werden. Das führt einerseits zu einer Überforderung der eigenen Inspektoren und andererseits zu immer mehr Rückfragen der Inspektoren bei Experten und bei den Herstellern.
All dies bindet unnötigen Ressourcen bei der FDA und verzögert die Zulassung der Medizinprodukte.
d) Welchen Nutzen bringt ASCA?
Somit soll das ASCA-Programm folgende Ziele erreichen:
- Patientensicherheit erhöhen
- Patientenversorgung durch schneller verfügbare Medizinprodukte verbessern
- Aufwände der Behörde und anderer Akteure (z. B. Hersteller) verringern
- Zulassungsverfahren beschleunigen
Wahrscheinlich ist der Nutzen für die FDA-Prüfer am höchsten. Diese müssen weniger lesen und weniger häufig Rücksprache mit ihren internen und externen Experten halten.
Die ASCA-akkreditierten Prüflabore dürften von steigenden Auftragszahlen profitieren.
Ob für Hersteller die schnelleren Zulassungszeiten die höheren Kosten aufwiegen, hängt vom Einzelfall ab.
2. Ablauf
a) Qualifizierung der Prüfhäuser
Die Prüfhäuser müssen sich bei einem „Accreditation Body“ akkreditieren. Dann können sie sich bei der FDA für das ASCA-Programm bewerben. Erst dann dürfen sie Produkte prüfen, die von der beschleunigten Zulassung profitieren (s. Abb. 1).
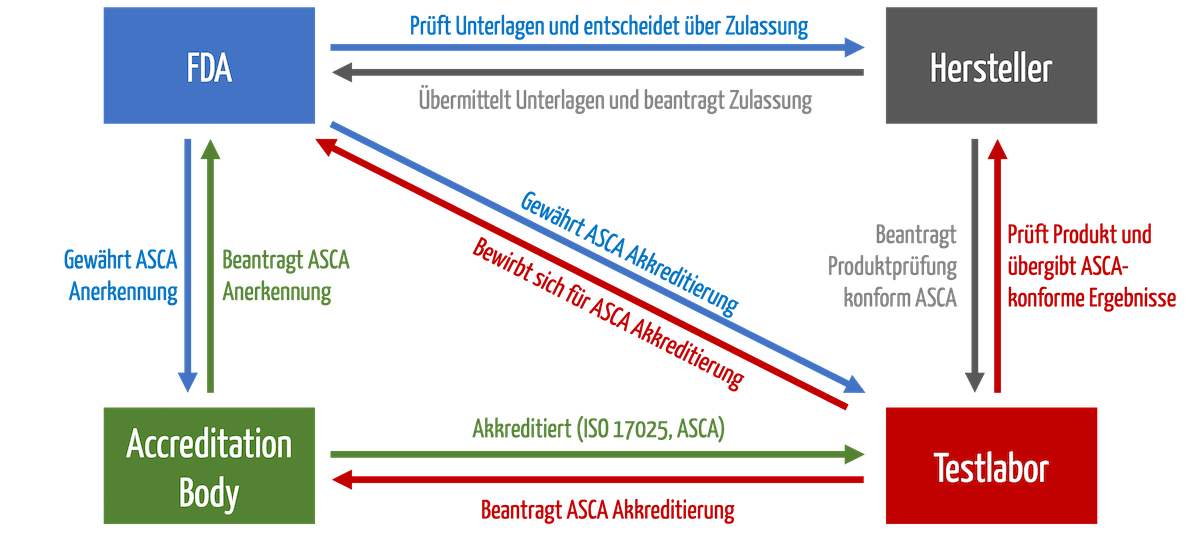
b) Zulassung der Produkte
Die Hersteller entwickeln ihre Produkte wie gewohnt. Bei der Zulassung gehen sie folgendermaßen vor:
- Regulatorische Strategie planen (z. B. Auswahl und Reihenfolge der Zielmärkte)
- Liste der Normen auswählen, mit denen Konformität erklärt werden soll (Diese enthält auch Normen, für die ASCA nicht angewendet werden soll oder kann.)
- Prüflabor auswählen (Eine Liste findet sich hier; in Deutschland gibt es aktuell ein akkreditiertes Prüflabor.)
- Mit dem Prüflabor einen geeigneten Testplan erstellen (Dieser beinhaltet die notwendigen und anwendbaren Leistungstests, die ebenfalls vom Prüflabor durchgeführt werden müssen, sowie die Testbedingungen, Ergänzungen oder Änderung von Tests, Akzeptanzkriterien gemäß Risikoakzeptanz.)
- Produkt prüfen und die Prüfergebnisse im ASCA-Prüfbericht durch das Labor zusammenfassen lassen
- Einreichungsunterlagen für die FDA zusammenstellen (Dazu zählen der „Cover letter“ mit Referenz zu ASCA-Tests, der Summary Test Report, die ASCA Declaration of Conformity sowie weitere Unterlagen.)
- Unterlagen bei der FDA einreichen (Bereits bei einer Beantragung über eStar kann man auswählen, dass die Prüfung gemäß ASCA erfolgt, s. Abb. 2.)

Dann ist es die Aufgabe der FDA, die Unterlagen zu prüfen. Bei der Zulassung durchläuft das Produkt die üblichen Phasen (s. Abb. 3).
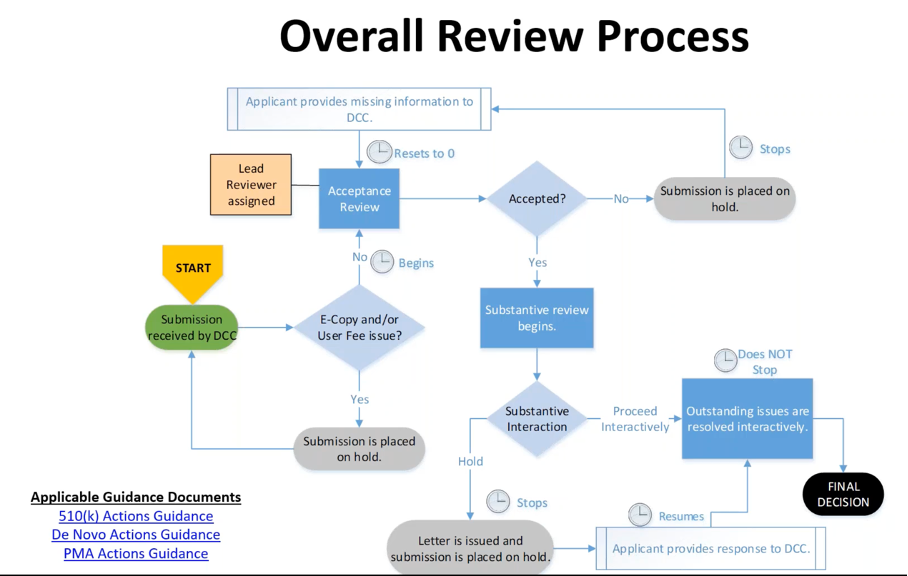
Die eigentliche Zeitersparnis betrifft die Schritte „Substantive Interaction“ und „Outstanding issues are resolved interactively“. Diese sollen durch das Programm verkürzt, die Iterationen nach Möglichkeit sogar vermieden werden.
Die FDA stellt auf ihrer Webseite umfangreiche Informationen zur Verfügung. Besonders relevant sind:
3. Anforderungen an die ASCA-Prüflabore
a) ISO/IEC 17025
Für Prüflabore gilt die QM-Norm ISO/IEC 17025. Die Konformität mit dieser Norm ist eine Voraussetzung. Die ASCA konkretisiert die Anforderungen dieser Norm.
Bezüglich Ausbildung und Kompetenz des Personals müssen ASCA-akkreditierte Prüfhäuser folgende Anforderungen erfüllen:
- Das technische Personal muss die Anforderungen der Normen (z. B. der IEC 60601-1) verstehen. Das betrifft insbesondere zugrunde liegende Konzepte wie die Basissicherheit und die wesentlichen Leistungsmerkmale.
- Das Labor ist zur Durchführung eines Programms für die Aus- und Weiterbildung des technischen Personals verpflichtet.
- Dieses Programm verpflichtet zu Schulungen und zur Dokumentation der Schulungen und ihrer Ergebnisse.
- Es muss fortlaufend Schulungen beinhalten (on the job oder Präsenztraining), die in bestimmten Abständen erfolgen oder/und wenn Prüfnormen oder -methoden aktualisiert oder neu entwickelt werden.
- Das Prüflabor muss bereits in den Stellenbeschreibungen die Anforderungsprofile genau spezifizieren.
b) Übersicht über die ASCA-Anforderungen
Die Anforderungen betreffen nicht nur die Kompetenz des Personals, sondern auch:
- Ausstattung des Prüflabors
- Verifizierungs- und Validierungsmethoden
- Erstellung und Lenkung von Testplänen und Prüfanweisungen
- Berücksichtigung der Zweckbestimmung und der beabsichtigten Verwendung des Produktes in den Testplänen
- Erstellung der Prüfberichte, insbesondere des zusammenfassenden ASCA-Prüfberichts
- Umfang und Inhalt der Prüfberichte (Beispielsweise muss das Prüflabor den Herstellern alle Meinungen und Interpretationen schriftlich mitteilen, einschließlich der Bedenken hinsichtlich der grundsätzlichen Sicherheit und der wesentlichen Leistungsmerkmale.)
c) Zusätzliche Anforderungen der FDA
Die FDA spezifiziert darüber hinaus Anforderungen an die Zulassungsdokumente, insbesondere:
- Cover Letter
- Declaration of Conformity
- ASCA Summary Test Report
- Prüfplan und -verfahren
- Abnahmekriterien und Ergebnisse, die den Sicherheitsanspruch begründen
4. Tipps und Hinweise
a) Produkt vorprüfen lassen
Der ASCA Summary Test Report wird ein zentrales Dokument, durch das die Prüfzeit erheblich verkürzt wird. Voraussetzung ist, dass der Report vollständig und korrekt ist. Der Summary Test Report wird auch Bedenken nennen, die das Prüfhaus hat, und erwähnen, ob ein Test wiederholt werden musste oder Änderungen am Produkt notwendig waren.
Daher kann es sinnvoll sein, das Produkt auch entwicklungsbegleitend testen zu lassen, um Überraschungen bei der ASCA-Prüfung zu vermeiden.
b) Präzises Sicherheitskonzept erstellen
Zu den häufig auftretenden Schwierigkeiten zählen eine ungenügend genaue oder gar fehlende Dokumentation der Sicherheitskonzepte bzw. der Systemarchitektur.
Eine Pauschalaussage wie „Das Produkt muss die Anforderungen der IEC 60601-1 erfüllen“ wird nicht mehr ausreichen. Benannte Stellen in Europa schauen heute schon genauer hin und die Prüflabore müssen es auch tun.
Das bedeutet: Hersteller sollten bereits während des Entwurfs die Sicherheitsanforderungen und Sicherheitsziele bezüglich der Erstfehlersicherheit spezifizieren und dann die Architektur als Vorgabe nutzen, um sichere Systeme zu bauen. Eine Hilfestellung kann die Grundnorm für funktionale Sicherheit IEC 61508 bieten.
Lesen Sie mehr zum Systems-Engineering und zum V-Modell als Dokumentationsmodell.
c) Akzeptanzkriterien spezifizieren
Die Prüfpläne sollten angemessene Testmethoden für die Leistungsprüfung mit genauen Akzeptanzkriterien spezifizieren. Sowohl die Hersteller als auch die Prüflaboren sollten beachten, dass Systeme ein Zeitverhalten haben. Dieses wird oft nicht geprüft.
Prüflabore sind dann in der Lage, schneller einen passenden Testplan zu erstellen und mit dem Hersteller einen angemessenen Versuchsaufbau zu definieren.
Weitere wichtige Informationen für das Prüflabor sind:
- Zweckbestimmung/bestimmungsgemäßer Gebrauch inklusive vorgesehener Nutzungsumgebung und vorgesehener Nutzer
- Umfassende Produktbeschreibung
- Systematische Herleitung der wesentlichen Leistungsmerkmale
- Diskussion der besonderen Eigenschaften der eingesetzten Technologien
- Informationen zur Kennzeichnung, insbesondere bezüglich Zweckbestimmung und Anwendung unter verschiedenen Umständen
- Nachweise zu den Leistungstests, wie Prüfberichte und klinische Daten
- Herleitung von Grenzwerten und Akzeptanzkriterien
- Beschreibung von Änderungen an Tests oder zusätzliche Tests, die notwendig sind
Diese Informationen können entweder in der Produktbeschreibung enthalten sein, im Architekturdokument oder in einzelnen Dokumenten. Eine Vorgabe für die Struktur gibt es nicht. Das ist allerdings nicht neu, denn all diese Angaben fordern Benannte Stellen und die FDA ohnehin schon.
d) Kosten beachten
Die FDA schätzt den zusätzlichen Aufwand der Prüflabore für das Erstellen der Berichte auf ca. 47 Stunden, also etwa eine Arbeitswoche. Das dürfte Kosten in der Größenordnung von 10.000 EUR nach sich ziehen.
5. Fazit und Zusammenfassung
a) Mögliche Nachteile
Die Prüflabore haben einen höheren Aufwand, was die Kosten nach oben treibt und dazu führen kann, dass kleinere Prüflabore an dem Programm nicht teilnehmen können. Das könnte eine eingeschränkte Prüfkapazität zur Folge haben.
Die Prüflabore werden die Kosten an die Hersteller weitergeben. Eine mögliche Folge ist, dass Firmen, die sich die Kosten nicht leisten können, nicht von den beschleunigten Verfahren der FDA profitieren können. Dann erreicht die FDA nicht ihr Ziel, innovative Medizinprodukte schneller in den Markt zu bringen.
b) Mögliche Vorteile
Präzise Anforderungen haben auch Vorteile:
- Sie ersparen allen Beteiligten (Herstellern, Prüfhäusern, Zulassungsstellen) unnötige Diskussionen.
- Hersteller wissen früh, was sie erwartet. Entsprechend früh können sie die Konformität sicherstellen, was teure Nachbesserungen und Iterationen erspart.
- Damit werden Prüf- und Zulassungsverfahren schneller und planbarer.
6. Updates zum ASCA-Programm
Die FDA hat im September 2024 ein Update zu dem Draft Guidance Basic Safety and Essential Performance of Medical Electrical Equipment, Medical Electrical Systems, and Laboratory Medical Equipment – Standards Specific Information for the Accreditation Scheme for Conformity Assessment (ASCA) Program veröffentlicht.
Lesen Sie das wichtigste in Kürze:
a) Vom Pilotprojekt zum Vollprogramm
Das ASCA-Programm hat sich von einem Pilotprojekt zu einem vollwertigen Programm entwickelt und Hersteller können freiwillig ein ASCA-akkreditiertes Prüflabor in Anspruch nehmen. Teilnehmen dürfen nur Prüflabore mit einer Akkreditierung durch eine US-amerikanische Stelle, während in Deutschland nur Prüflabore teilnehmen können, die auch eine internationale Akkreditierung besitzen. Prüfberichte von durch die Deutsche Akkreditierungsstelle (DAkkS) akkreditierten Laboren werden weiterhin von der FDA anerkannt.
Hier finden Sie eine Liste der derzeit akkreditierten Prüflabore.
ASCA-Prüflabore werden regelmäßig nach erweiterten Anforderungen, die über ISO/IEC 17025 hinausgehen, geprüft, um ihre Kompetenz sicherzustellen.
b) Erweiterte Zusammenarbeit und Prüfstandards
Prüflabore werden enger mit Herstellern zusammenarbeiten, um Testpläne zu erstellen. Dabei liegen die Schwerpunkte auf den beiden Themen elektromagnetische Verträglichkeit (EMV) und wesentliche Leistungsmerkmale. Der aktualisierte Draft Guidance betont die wesentlichen Leistungsmerkmale eines Geräts, die erhalten bleiben müssen, um Risiken für Patienten oder Anwender zu vermeiden. Prüflaboratorien müssen dieses Konzept verstehen und eng mit den Herstellern zusammenarbeiten, um die Einhaltung dieser Anforderungen sicherzustellen. Die Testberichte müssen auch auf Probleme hinweisen, die während dem Test aufgetreten sind.
Hersteller können künftig davon ausgehen, dass ASCA-Prüflabore nicht nur die Risikomanagementakte formal prüfen, sondern auch deren inhaltliche Konsistenz bewerten und bei Bedarf zusätzliche Tests planen. Es bleibt zu hoffen, dass Benannte Stellen die Bewertungen der ASCA-Prüflabore anerkennen. Aktuell erleben wir jedoch häufig, dass Benannte Stellen von den Laborbewertungen abweichen und eigene Abweichungen festhalten.
c) Warum diese Änderungen wichtig sind
Der Fokus der Prüflabore lag bisher stark auf dem Bereich der Basissicherheit. Mit dem ASCA-Programm treibt die FDA den Fokus in Richtung der Funktionssicherheit von elektrisch betriebenen Medizinprodukten (IVD eingeschlossen). Das bedeutet, dass Hersteller die Risiken vollständig ermitteln und die Funktionssicherheit in der Architektur oder in einem Sicherheitskonzept nachvollziehbar beschreiben.
Die Anforderungen an den Nachweis der Sicherheit von Medizinprodukten werden in Zukunft auch unabhängig vom ASCA-Programm strenger. Eine Arbeitsgruppe, der wir angehören, entwickelt derzeit den technischen Report IEC TR 60601-4-9 „Medizinische elektrische Geräte – Teil 4-9: Leitfaden und Interpretation – Funktionssicherheit bei wesentlichen Leistungsmerkmalen“. Der Report befindet sich aktuell im Entwurf und soll 2025 veröffentlicht werden. Spätestens dann werden Prüflabore die Funktionssicherheit von Medizinprodukten intensiver prüfen.
Nutzen Sie die Hilfe des Johner Instituts, um mit präzisen Unterlagen und sicheren Medizinprodukten alle Zulassungsverfahren schnell und mit minimalen Aufwänden zu durchlaufen.
Wir unterstützen Sie bei der Erstellung Ihrer Unterlagen (z. B. Sicherheitskonzept, Risikoanalyse, Festlegung der wesentlichen Leistungsmerkmale, Testpläne, Prüfstrategie) oder nehmen Ihnen diese Arbeit ganz ab.
Unsere Expert:innen begleiten Sie auch beim Review aller Unterlagen wie der Architektur (konform mit ISH1, mit Methoden der funktionalen Sicherheit) und aller anderen „Submission-Dokumente“.
Änderungshistorie
- 2024-11-25: Kapitel 6 mit Updates zum ASCA-Programm ergänzt
- 2023-06-20: Artikel initial erstellt


