Hersteller müssen die biologische Sicherheit und damit die Biokompatibilität ihrer Medizinprodukte nachweisen. Dabei hilft die Normenfamilie ISO 10993.
Inhalt
Diese Seite verschafft eine Übersicht über:
- Biologische Sicherheit und Biokompatibilität: Beispiele, Definitionen
- Regulatorische Anforderungen an die biologische Sicherheit
- Ansatzpunkte zum Erreichen der biologischen Sicherheit / Biokompatibilität
- Nachweise der biologischen Sicherheit / Biokompatibilität (Relevanz von Tests)
- Unterstützung für Medizinproduktehersteller
1. Biologische Sicherheit und Biokompatibilität
1.1 Beispiele
1.1.1 Beispiele für biologische Gefährdungen und Risiken
Medizinprodukte, die physischen Kontakt, insbesondere mit Patienten, haben, bergen biologische Risiken, die beispielsweise verursacht werden durch:
- Chemikalien, die sich aus Materialien des Medizinprodukts herauslösen,vom Körper aufgenommen werden und gesundheitsbedenkliche Auswirkungen haben
- Implantate, die eine Abstoßungsreaktion hervorrufen
- Materialien, die eine allergische Reaktion verursachen
- Materialien, die entgegen der Absicht des Herstellers vom Körper abgebaut oder umgewandelt werden
- Implantate und Materialien, die nicht wie gewünscht in den Körper einwachsen
1.1.2 Beispiele für Schäden
Diese Risiken können zu vielerlei Schäden führen:
- Rötungen, Schwellung, lokale Entzündung
- Allergien
- Fieber
- Systemische Toxizität z.B. Organschäden, Vergiftung
- Krebs
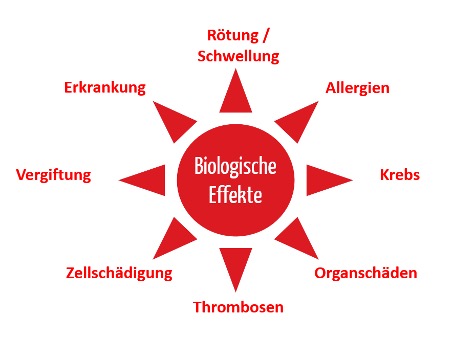
Abb. 1: Beispiele für Schäden bei mangelnder biologischer Sicherheit
1.2 Definitionen
1.2.1 Biologische Risiko
Die ISO 10993-1 definiert den Begriff biologisches Risiko wie folgt:
„Kombination der Wahrscheinlichkeit von gesundheitlichen Schäden aufgrund unerwünschter Reaktionen im Zusammenhang mit dem Medizinprodukt oder Wechselwirkungen mit den Materialien und der Schwere dieser Schäden.“
Quelle: ISO 10993-1
1.2.2 Biologische Sicherheit
Damit versprichtdie biologische Sicherheit die Freiheit von nicht akzeptablen biologischen Risiken im Zusammenhang mit der bestimmungsgemäßen Verwendung.
Biokompatibilität
Etwas kryptischer wirkt die Definition des Begriffs „Biokompatibilität“:
„Fähigkeit eines Medizinprodukts oder Materials, mit einer angemessenen Host-Reaktion Leistung in einer spezifischen Anwendung zu erbringen“
Quelle: ISO 10993-1
Der Begriff Host-Reaktion meint hier mögliche unerwünschte und nachteilige Reaktionen. Die Norm merkt an, dass der Nachweis der Biokompatibilität durch biologische Prüfungen und durch eine Beurteilung der herauslösbaren Chemikalien erfolgen kann.
In der biologischen Risikobeurteilung nach ISO 10993-1 wird die biologische Sicherheit unter Betrachtung der biologischen Risiken bewertet, die verbunden sind mit:
- den Bestandteilen eines Medizinprodukts und
- den Wechselwirkungen zwischen Produkt und Gewebe
Die biologischen Risken in Zusammenhang mit Infektionserregern fallen nicht in den Anwendungsbereich der ISO 10993!
1.3 Endpunkte der biologischen Sicherheit nach ISO 10993
Hersteller müssen die Biokompatibilität ihrer Medizinprodukte gewährleisten und daher die Wahrscheinlichkeit von Schäden aufgrund mangelnder biologischer Sicherheit minimieren.
Die spezifischen (negativen) biologischen Effekte, welche die Hersteller bei der biologischen Bewertung überprüfen müssen, nennt man Endpunkte. Beispiele für Endpunkte sind:
- Zytotoxizität (Zellschädigung)
- Sensibilisierung der Haut (z. B. allergische Reaktion)
- Irritation der Haut oder des Gewebes (z. B. Rötung und Juckreiz)
- Systemische Toxizität: akute, subakute, subchronische und chronische Toxizität (z. B. Organschäden, Tod)
- Materialbedingte Pyrogenität (Hervorrufenvon Fieber)
- Genotoxizität (Schädigung der DNA)
- Karzinogenität (Verursachen von Krebs)
- Reproduktionstoxizität (Beeinträchtigung der Fortpflanzungsfähigkeit)
- Implantationseffekte (z. B. Abstoßung von Implantaten)
- Blutunverträglichkeit (z. B. Thrombose)
Der Artikel beschreibt in den Abschnitten 4 und 5, wie Hersteller die biologische Sicherheit erreichen und überprüfen können.
2. Regulatorische Anforderungen
2.1 Die Anforderungen der MDR
Die Medizinprodukteverordnung (Medical Device Regulation MDR) fordert die Biokompatibilität von Medizinprodukten an mehreren Stellen:
Anhang I: Grundlegende Sicherheits- und Leistungsanforderungen
Der Anhang I der MDR erwähnt den Begriff Biokompatibilität nicht explizit. Er schreibt aber:
Die Produkte werden so ausgelegt, hergestellt und verpackt, dass die Risiken durch Schadstoffe und Rückstände für Patienten — unter Berücksichtigung der Zweckbestimmung des Produkts — sowie für Transport-, Lager- und Bedienungspersonal so gering wie möglich gehalten werden. Dabei wird Geweben, die diesen Schadstoffen und Rückständen ausgesetzt sind, sowie der Dauer und Häufigkeit der Exposition besondere Aufmerksamkeit gewidmet.
MDR, Anhang I, Absatz 10.2
Bereits im ersten Abschnitt des Anhangs I fordert die Verordnung, die Sicherheit und Gesundheit von Patienten, Anwendern und Dritten nicht zu gefährden und die „Risiken durch sichere Auslegung und Herstellung beseitigen oder so weit wie möglich [zu] minimieren“.
Anhang II: Technische Dokumentation
Die MDR verpflichtet die Hersteller im Anhang II, Informationen zu allen Prüfungen zu dokumentieren inklusive
Testaufbau, Testprotokolle, Methoden der Datenanalyse, […], Testergebnisse hinsichtlich der Biokompatibilität des Produkts einschließlich der Identifizierung aller Materialien in direktem oder indirektem Kontakt mit dem Patienten oder Anwender, [sowie hinsichtlich der …] chemischen und mikrobiologischen Parameter.
MDR, Anhang II, Absatz 6.1.b)
Auch den Fall, dass die Hersteller keine Tests durchführen, regelt die MDR:
Falls keine neuen Tests durchgeführt wurden, wird diese Entscheidung in der Dokumentation begründet. Eine solche Begründung wäre beispielsweise, dass Biokompatibilitätstests an identischen Materialien durchgeführt wurden, als diese Materialien in ein rechtmäßig in Verkehr gebrachtes oder in Betrieb genommenes Vorgängermodell des Produkts integriert wurden.
MDR, Anhang II, Absatz 6.1.b)
Anhang VII: Anforderungen an Benannte Stellen
Dieser Anhang betrifft die Hersteller zumindest indirekt, denn darin verpflichtet die MDR die Benannten Stellen:
- Die Kompetenz des Personals der Benannten Stellen bezüglich der Biokompatibilität gewährleisten
- Aktualität des Wissens der Hersteller
Benannte Stellen müssen die Verfahren der Hersteller prüfen, mit denen die Hersteller sicherstellen, dass sie über neuste wissenschaftliche Erkenntnisse auch in Bezug auf die Biokompatibilität von Materialien verfügen.
2.2 Die Anforderungen der FDA
2.2.1 ISO 10993 als Recognized Consensus Standards
Die FDA erwartet ebenfalls die biologische Sicherheit von Medizinprodukten. Sie zählt die Normenfamilie ISO 10993 zu den Recoginized Consensus Standards.
2.2.2 Guidance Document zur ISO 10993-1
Zudem hat die FDAein ausführliches Guidance-Dokument zur ISO 10993-1 für die Bewertung der Biokompatibilität publiziert. Darin beschreibt sie sowohl das Vorgehen nach ISO 10993-1 im Detail als auch zusätzliche Anforderungen wie weitere Endpunkte für bestimmte Produktkategorien. Die fehlenden Endpunkte in der ISO 10993-1 sind ein Grund, warum die ISO 10993-1:2018 nur teilweise von der FDA anerkannt wird.
2.2.3 Guidance Document zu Biokompatibiltätsnachweisen ohne Tierversuche
In der 2023 veröffentlichten Version des Dokuments wurde der Anhang G „Biocompatibility of Certain Devices in Contact with Intact Skin“ hinzugefügt, um den „3R-Ansatz“ zu Reduzierung, Verbesserung und Ersatz von Tierversuchen weiter zu unterstützen.
2.2.4 Entwurf des Guidance Documents zu chemischen Analysen und ISO 10993-18
Im September 2024 hat die FDA zudem einen neuen Entwurf eines Guidance-Dokuments zur Biokompatibilität veröffentlicht, dass sich speziell mit der Durchführung chemischer Analysen befasst.
- Ziel: Ziel des Dokuments ist die Standardisierung der Vorgehensweise. Es schließt damit den Spielraum, den die Norm lässt.
- Fokus: Der Fokus der Guidance liegt auf Extractables-Studien, also der Identifizierung und Quantifizierung von Substanzen, die nach der Extraktion des Medizinprodukts in Lösungsmitteln freigesetzt werden.
- Inhalt: Die Guidance bietet detaillierte Empfehlungen zur Durchführung der Extraktion, zur chemischen Analytik sowie zur Notwendigkeit einer umfassenden und präzisen Dokumentation – ergänzend zur ISO 10993-18.
- Zielgruppe: Das Dokument wendet sich v. a. an Toxikologen, Biokompatibilitätsbewerter und Experts für toxikologische Risikobewertungen, aber vor allem auch Labore, die chemische Analysen durchführen.
- Kritik: Kritisch anzumerken ist, dass die Anforderungen an den Umfang der chemischen Analysen, die Datenqualität und insbesondere die Dokumentation sehr hoch sind. Dies dient der Erhöhung der Produktsicherheit, könnte jedoch in einem unverhältnismäßigen Anstieg des Aufwands und der Kosten für Laborprüfungen resultieren – selbst bei bewährten Standardmaterialien.
Fazit: Die Anforderungen an die Biokompatibilität steigen. Daher ist es wichtig, die Laboruntersuchungen sehr gezielt auszuwählen und durchzuführen.
2.3 Die Anforderungen der Normenfamilie ISO 10993
2.3.1 Übersicht über die Normenfamilie
Die Normenfamilie 10993 zur „Biologischen Sicherheit von Medizinprodukten“ besteht aus mehreren Teilen, die teilweise unter der MDR harmonisiert sind. Die Harmonisierung von Normen ist ein fortlaufender Prozess, so dass sich der Status ändert. Es ist zu empfehlen regelmäßig die offiziellen Veröffentlichungen der Europäischen Kommission und des Europäischen Komitees für Normen (CEN) zu konsultieren, um den aktuellen Stand der harmonisierten Normen zu überprüfen.
| Norm |
Titel |
Harmonisiert unter MDR? |
| ISO 10993-1 |
Beurteilung und Prüfungen im Rahmen eines Risikomanagementprozesses |
Nein |
| ISO 10993-2 |
Tierschutzbestimmungen |
Nein |
| ISO 10993-3 |
Prüfungen auf Gentoxizität, Karzinogenität und Reproduktionstoxizität |
Nein |
| ISO 10993-4 |
Auswahl von Prüfungen zur Wechselwirkung mit Blut |
Nein |
| ISO 10993-5 |
Prüfungen auf In-vitro-Zytotoxizität |
Nein |
| ISO 10993-6 |
Prüfungen auf lokale Effekte nach Implantationen |
Nein |
| ISO 10993-7 |
Ethylenoxid-Sterilisationsrückstände |
Nein |
| ISO 10993-9 |
Rahmen zur Identifizierung und Quantifizierung von möglichen Abbauprodukten |
Ja |
| ISO 10993-10 |
Prüfungen auf Hautsensibilisierung |
Ja |
| ISO 10993-11 |
Prüfungen auf systemische Toxizität |
Nein |
| ISO 10993-12 |
Probenvorbereitung und Referenzmaterialien |
Ja |
| ISO 10993-13 |
Qualitativer und quantitativer Nachweis von Abbauprodukten in Medizinprodukten aus Polymeren |
Nein |
| ISO 10993-14 |
Qualitativer und quantitativer Nachweis von keramischen Abbauprodukten |
Nein |
| ISO 10993-15 |
Qualitativer und quantitativer Nachweis von Abbauprodukten aus Metallen und Legierungen |
Ja |
| ISO 10993-16 |
Entwurf und Auslegung toxikokinetischer Untersuchungen hinsichtlich Abbauprodukten und herauslösbaren Bestandteilen |
Nein |
| ISO 10993-17 |
Toxikologische Risikobewertung von Medizinproduktbestandteilen |
Ja |
| ISO 10993-18 |
Chemische Charakterisierung von Werkstoffen für Medizinprodukte im Rahmen eines Risikomanagementsystems |
Ja |
| ISO 10993-19 |
Physikalisch/chemische, mechanische und morphologische Charakterisierung |
Nein |
| ISO 10993-20 |
Prinzipien und Verfahren für die immuntoxikologische Prüfung von Medizinprodukten |
Nein |
| ISO 10993-23 |
Prüfungen auf Irritation |
ja |
2.3.2 Die Basisnorm ISO 10993-1
Die Basisnorm ISO 10993-1 legt auch die Endpunkte entsprechend der Produktk ategorisierung nach Kontaktart und Dauer fest, die die Hersteller bei der Bewertung der biologischen Sicherheit adressierenmüssen.
Diese Endpunkte beschreibt die Norm in Tabelle A.1 im Annex A. Der folgende Ausschnitt aus der Tabelle zeigt exemplarisch die Endpunkte für ein Medizinprodukt mit Kontakt zu zirkulierendem Blut.

Abb. 2: Entnommen aus der ISO 10993-1 Annex A, Tabelle A.1
Die Tabelle stellt keine Checkliste für Prüfungen dar, sondern gibt Hilfestellung welche Endpunkte für die Produktkategorie bewertet werden sollen.
Wichtig ist auch, dass gemäß ISO 10993-1 ausdrücklich eine Materialcharakterisierung durchzuführen und auf Tierversuche zu verzichten ist. Zudem ermöglicht eine Materialcharakterisierung eine höhere Aussagekraft und damit eine höhere biologische Sicherheit.
Die ISO 10993-1 stellt klar, dass die Hersteller die biologische Sicherheit über den kompletten Lebenszyklus des Medizinprodukts gewährleisten müssen:
„The biological safety of a medical device shall be evaluated by the manufacturer over the whole life cycle of a medical device.“
ISO 10993-1, 4.7
Ebenso ergänzt dieser Teil die Forderungen nach „Freiheit von inakzeptablen biologischen Risiken“ wie folgt:
„The following shall be taken into account for their relevance to overall biological evaluation of the medical device: …process contaminants and residues… packaging materials… leachables substances… degradation products…“
ISO 10993-1, 4.3
2.4 Weitere regulatorische Anforderungen
Neben der Normenfamilie ISO 10993 sollten Hersteller neben Produktspezifischen Normenweitere regulatorische Vorgaben beachten wie beispielsweise:
3. Biologische Sicherheit erreichen
Biologische Sicherheit zu erreichen, bedeutet die biologischen Risiken zu minimeren. Die MDR und ISO 14971 beschreiben drei Typen an risikominimierenden Maßnahmen:
- Maßnahmen, die inhärente Sicherheit schaffen
- Schutzmaßnahmen
- Maßnahmen in Form von Informationen
Maßnahmen zur biologischen Sicherheit sind:
- Hochwertige und für die Anwendung geeignete Materialien verwenden
- Saubere Produktion gewährleisten
- Produkte mit geeigneten Verfahren reinigen, desinfizieren, sterilisieren
- Kontakt mit dem Produkt vermeiden (z.B. durch Schutzausrüstung) oder reduzieren (z.B. verkürzen)
4. Biologische Sicherheit nachweisen
4.1 Nachweise mit Fokus auf die Medizinprodukte
Die Hersteller sind verpflichtet, die Wirksamkeit dieser Maßnahmen zu überprüfen und damit die biologische Sicherheit bzw. Biokompatibilität bezüglich der oben genannten Endpunkte (soweit relevant) nachzuweisen.
Als ersten Schritt werden Informationen zum zu bewertenden Medizinprodukt für die Nachweisführung zusammengetragen.
- Materialdatenblätter wie zum Beispiel Sicherheitsdatenblätter zu den Rohstoffen, Technische Datenblätter zur Verarbeitung und Eignung
- Zertifikate z.B. hinsichtlich Biokompatibilität des eingekauften Materials
- Ergebnisse von Recherchen zu bekannten Inhaltstoffen und Additiven
- Verwendete Produktionshilfsmittel
- Markterfahrung
VORSICHT: Die Informationen sind notwendig und wichtig, aber meist nicht hinreichend, um die Biokompatibilität des finalen Produktes nachzuweisen.
Es gibt zusätzliche Einflüsse auf die Biokompatibilität des Produkts, die betrachtet werden müssen, um alle notwendigen Endpunkte bewerten zu können:
- Verarbeitung des Materials z.B. Polymerisation
- Verwendete Additive und Farbstoffe neben den Hauptbestandteilen
- Klebeprozesse und Wechselwirkungen
- Lasergravur und Beschriftungen
- Endreinigung und verwendete Prozesschemikalien
- Sterilisation und Materialeinfluss
- Verpackung und Lagerung

Abb. 3: ISO 10993 – Exemplarische Einflussfaktoren auf die Biokompatibilität
FAZIT: Der Nachweis der Biokompatibilität lässt sich nicht ausschließlich anhand von Datenblätter zum Material führen. Daher sind meistens Prüfungen am finalen Produkt notwendig.
Die Normenfamilie ISO 10993 beschreibt, wie die Nachweise erfolgen sollten. Beispielsweise sollte nach der gut geplanten Chemischen Charakterisierung nach ISO 10993-18 die toxikologische Bewertung nach ISO 10993-17 der unter den schlechtesten Bedingungen bezüglich der Exposition erfolgen. Dabei prüfen die Hersteller das finale Produkt, ob es frei von biologischen Gefährdungen ist wie z.B. von toxikologisch relevanten Stoffen (organisch und/oder anorganisch) ist..
Bei den Nachweisen sollten die Hersteller möglichst auf Tierversuche verzichten. Um die Kosten zudem gering zu halten und eine erfolgreiche toxikologische Bewertung der Ergebnisse überhaupt zu ermöglichen, ist eine risikobasierte und gut geplante Prüfstrategie sowie das Festlegen von anwendungsabhängigen Prüfparametern notwendig.
4.2 Nachweise mit Fokus auf die Prozesse
- Die biologische Sicherheit von Medizinprodukten umfasst neben den Aspekten der Biokompatibilität der verwendeten Materialien auch mögliche Gefährdungen durch infektiöse Mikroorganismen. Um biologische Risken durch oberflächliche Kontaminationen mit z.B. pathogenen Bakterien/Viren zu minimieren, werden Prozesse eingeführt, die validiert sein müssen.
Dazu zählen
5. Unterstützung
Haben Sie noch Fragen zur Biokompatibilität oder der Normenfamilie ISO10993? Dann nutzen Sie das kostenfreie Micro-Consulting.
Falls Sie sich tiefer in das Thema einarbeiten wollen, dann bieten sich an:
- Das Grundlagenseminar Biokompatibilität
- Ein umfangreicher Selbstlernkurs in der Johner Academy
Melden Sie sich, wenn Sie Unterstützung benötigen, bei
- Validierung der Prozesse wie z.B. Aufbereitung, Endreinigung und/oder Sterilisation
- der Entwicklung ihrer biokompatiblen Medizinprodukte,der Erstellung der Prüfstrategie zur Biokompatibilität und der Auswahl eines Prüflabors sowie Organisation der notwendigen Prüfungen
- Toxikologischer Risikobewertung der Ergebnisse,
dem Erstellen einer schlanken und normenkonformen Technischen Dokumentation.Damit stellen Sie sicher, dass Sie Ihre Produkte schnell und sicher entwickeln und ohne Probleme bei der Zulassung in den Markt bekommen.


