
Die Norm ISO 10993-17 ist Teil der Normenreihe ISO 10993 zur Biokompatibilität. Die ISO 10993-17 beschreibt die Verfahren der toxikologischen Risikobewertung.
Im Herbst 2023 erschien nach über 20 Jahren eine umfangreiche Überarbeitung der Norm.
Medizinproduktehersteller sollten wissen,
- wann sie die ISO 10993-17 erfüllen müssen,
- was die Norm verlangt,
- worin die aktuellen Änderungen bestehen sowie
- wann und wie sie diese Änderungen umsetzen sollten.
Dabei hilft dieser Artikel.
1. Kontext der ISO 10993-17
1.1 Das Konzept der toxikologischen Risikobewertung
Die toxikologische Risikobewertung (Toxicological Risk Assessment, TRA) ist ein wesentlicher Bestandteil der biologischen Sicherheitsbewertung. Sie dient dazu, potenzielle Gesundheitsrisiken zu identifizieren und diese anhand von Grenzwertermittlung und Expositionsabschätzung zu bewerten.
Gesundheitsrisiken können entstehen durch
- herauslösbare Substanzen (aus dem Material selbst),
- Verunreinigungen (beispielsweise durch die Produktion des Medizinprodukte) oder
- Abbauprodukte,
die vom Medizinprodukt in den menschlichen Körper gelangen.
Die toxikologische Risikobewertung ist eine Voraussetzung innerhalb der Bewertung der Biokompatibilität nach ISO 10993-1, um die gesetzlich geforderte Patientensicherheit bei der Anwendung von Medizinprodukten zu gewährleisten.
Der Artikel zu Biokompatibilität und ISO 10993-1 verschafft Ihnen einen leichten Einstieg in das Thema.
1.2 Die regulatorischen Anforderungen
Die gesetzlichen Anforderungen formuliert die MDR folgendermaßen:
Die Produkte werden so ausgelegt, hergestellt und verpackt, dass die Risiken durch Schadstoffe und Rückstände für Patienten — unter Berücksichtigung der Zweckbestimmung des Produkts — sowie für Transport-, Lager- und Bedienungspersonal so gering wie möglich gehalten werden. Dabei wird Geweben, die diesen Schadstoffen und Rückständen ausgesetzt sind, sowie der Dauer und Häufigkeit der Exposition besondere Aufmerksamkeit gewidmet.
MDR, Anhang I, Absatz 10.2
Die ISO 10993-1:2025 bezieht sich mehrmals auf die Notwendigkeit der toxikologischen Risikobewertung zur Beurteilung der chemischen Information hinsichtlich spezifischer Endpunkte wie z.B. Systemische Toxizität:
Systemic toxicity can be evaluated in a toxicological risk assessment of chemical information. Any toxicological risk assessment shall be conducted in accordance with the requirements of ISO 10993-17 unless otherwise justified.
ISO 10993-1:2025, Abschnitt 6.5.5
1.3 Begriffe und Abkürzungen
Dieser Artikel verwendet viele Konzepte und Definitionen der ISO 10993-17. Hier eine Übersicht:
| TCL | Tolerable Contact Level, tolerierbare Kontaktmenge | Abschätzung der Oberflächenkontakt-Exposition mit einem identifizierten Bestandteil, der keine nennenswerte Reizung hervorruft |
| TI | Tolerable Intake, tolerierbare Aufnahmemenge | Abschätzung der täglichen Exposition mit einem identifizierten Bestandteil über einen bestimmten Zeitraum (z. B. akut, subakut, subchronisch oder chronisch) auf der Grundlage der Körpermasse, die als gesundheitlich unbedenklich gilt |
| POD | Point of Departure | Niedriger Punkt auf einer toxikologischen Dosis-Wirkung-Kurve, die auf der Grundlage von Versuchs- oder Beobachtungsdaten erstellt wurde. Als POD kommen verschiedenen Werte in Betracht, wie der Wert der niedrigen Referenzdosis, der geringsten beobachteten schädlichen Wirkungen, der minimaler Reizung oder ein Wert ohne Reizung oder ohne beobachtete schädliche Wirkung. |
| EEDmax | Worst-Case-Expositionsdosis pro Kilogramm Körpergewicht | Die Expositionsdosis beschreibt die Menge eines Bestandteils, die über einen Expositionsweg während eines bestimmten Zeitraums mit dem Körper in Berührung kommt/kommen kann. |
| MoS | Margin of Safety, Sicherheitsspanne | Verhältnis zwischen der tolerierbaren Kontaktmenge des Bestandteils (Zähler) bzw. der tolerierbaren Aufnahme (Zähler) und der Expositionsdosis (Nenner) |
| TTC | Threshold of toxicological concern, toxikologisch bedenklicher Schwellenwert | Expositionsniveau, unterhalb dessen kein nennenswertes Risiko für die menschliche Gesundheit besteht |
1.4 ISO 10993-17 kaufen
Die deutsche Version der Norm kann man beim Beuth-Verlag für etwa 200 EUR erwerben.
Die englische Version der ISO 10993-17:2023 finden Sie für ein Zehntel der Kosten auf den Seiten von evs.ee.
2. Ablauf einer toxikologischen Risikobewertung
2.1 Übersicht
Vor der eigentlichen toxikologischen Risikobewertung (TRA) muss der Hersteller die Bestandteile identifizieren, mit denen der Patient während der klinischen Anwendung exponiert werden kann. Dabei sollte der Hersteller gemäß den Anforderungen aus der Schwesternorm ISO 10993-18 vorgehen.
Erst dann erfolgt die eigentliche TRA (Scope der ISO 10993-17), die sich in drei Schritte unterteilen lässt (s. Abb. 1):
- Identifikation von Gefährdungen, die sich aus den identifizierten Bestandteilen des Medizinprodukts ergeben können
- Risikoabschätzung inklusive
- Bestimmung der tolerierbaren Kontaktmenge oder Aufnahme (Alternativ: toxikologisch bedenklicher Schwellenwert: TTC)
- Abschätzung der Expositionsdosis (Worst-Case-Ansatz)
- Ableitung der Sicherheitsspanne (Margin of Safety, MoS)
- Risikobewertung: Der letzte Schritt ist die Bewertung des toxikologischen Risikos auf der Grundlage der identifizierten Gefährdungen, der tolerierbaren Kontaktmenge oder der tolerierbaren Aufnahme und der geschätzten Expositionsdosis für jeden Bestandteil. Die Risikoabschätzungen werden mit den Akzeptanzkriterien verglichen, um mögliche Lücken bzw. Restrisiken abzuleiten.
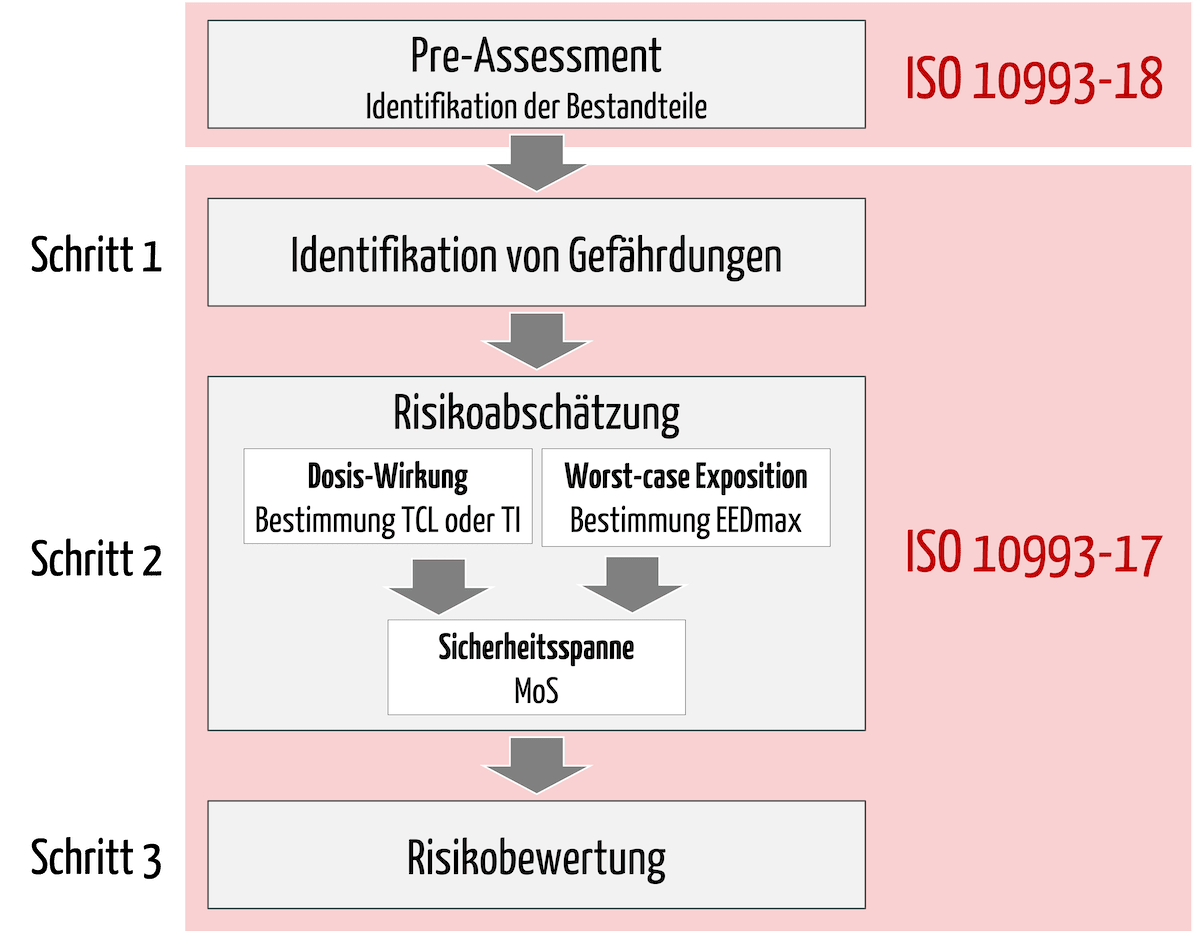
Bei allen Schritten der TRA sollten die Hersteller die Charakteristika des zu bewertenden Medizinprodukts und dessen klinische Anwendung berücksichtigen:
- Verwendete Materialien
- Anwendungsart, z. B. kumulierte Anwendung mit neuem Produkt
- Kontaktart der Exposition, z. B. Haut
- Expositionsdauer, z. B. chronisch
- Patientengruppe, z. B. Kinder
- Dimension/Kontaktfläche und Anzahl der Medizinprodukte in Anwendung
- Unsicherheitsfaktoren für die Verwendung der Daten aus der Literatur
2.2 Pre-Assessment: Identifikation der Bestandteile
Um toxikologische Gefährdungen einzuschätzen, muss der Hersteller die Bestandteile identifizieren, die während der klinischen Anwendung seines Medizinprodukts freigesetzt werden können. Dies erfolgt über eine umfangreiche Materialcharakterisierung der Bereiche des Medizinprodukts mit direktem und/oder indirektem Patientenkontakt.
In der Regel fehlen Informationen zu den Bestandteilen der verwendeten Materialien. Dann muss am Endprodukt eine chemische Analyse nach ISO 10993-18 durchgeführt werden. Durch die Wahl eines geeigneten Extraktionsverfahrens wird die klinische Anwendung simuliert, z. B. über Berücksichtigung der Kontaktoberflächen, Kontaktart sowie Kontaktdauer.
Die richtige Auswahl der Methodik in der chemischen Charakterisierung ist essenziell für eine erfolgreiche toxikologische Risikobewertung!
100 % Lösungsmittel, 72 h Extraktion bei 50 °C ist keine adäquate Simulation einer Kurzzeit-Anwendung eines Polymers auf intakter Haut.
Eine falsche Auswahl des Extraktionsmediums, der Dauer oder der Temperatur kann zur Degradation des Materials führen, die in der klinischen Anwendung nicht stattfindet.
Eine große Anzahl an nicht identifizierbaren Substanzen erschwert das TRA und kann zu Restrisiken führen, die weitere Prüfungen nach sich ziehen können.
2.3 Die eigentliche TRA
Schritt 1: Identifikation von Gefährdungen
Nachdem der Hersteller die Bestandteile kennt, die freigesetzt werden können, muss er im ersten Schritt bewerten:
- Müssen diese Bestandteile für die klinische Anwendung des Produkts betrachtet werden?
- Gehen von ihnen Gefährdungen aus?
Die neue Version der Norm erlaubt es, für diese Bewertung ein Toxicological Screening Limit (TSL) anzuwenden. Ein TSL wird verwendet, um die kumulative Expositionsdosis eines Bestandteils über einen bestimmten Zeitraum zu bewerten und festzustellen, ob sie ein vernachlässigbares toxikologisches Risiko darstellt. Liegt die maximale Expositionsdosis unter dem angegebenen TSL-Wert, ist eine weitere Risikobewertung für systemische Schäden nicht notwendig.
Wenn 100 μg eines identifizierten Bestandteils aus einem einzelnen Medizinprodukt pro Tag extrahiert werden und ein einzelnes Medizinprodukt weniger als oder gleich 30 Tage lang mit dem Körper in Kontakt ist (keine wiederholte Anwendung), ergibt sich für das TSL gemäß Tabelle B.1 der Norm:
TSL = 120 x 1 = 120 µg
Die kumulative Expositionsdosis (bei einmaliger Anwendung eines Produkts) entspricht den detektierten 100 µg.
Damit ist die kumulative Expositionsdosis kleiner als das TSL. Eine weitere Risikobewertung für systemische Schäden ist nicht notwendig.
Der TSL-Ansatz ist nicht vorgesehen für
- die Kohorte der besorgniserregenden Stoffe,
- nicht ausreichend identifizierte oder irritierende Bestandteile,
- empfindliche Patientenpopulationen (junge Säuglinge, d. h. sechs Monate oder jünger).
Zur Identifikation der Gefährdungen, die von den Freisetzungsprodukten aus dem Medizinprodukt ausgehen, erfolgt zunächst eine Sammlung und Evaluierung toxikologischer Daten zur Substanz (Datenbankenrecherche). Dabei sind die Berücksichtigung der klinischen Anwendung und die Qualität der Daten wichtig:
- Berücksichtigung der identifizierten Endpunkte anhand Kategorisierung des Medizinprodukts entsprechend ISO 10993-1
- Berücksichtigung des Expositionswegs, z. B. Haut, orale Aufnahme, Inhalation
- Bewertung der Qualität der Daten durch z. B. Angabe der Richtlinie, nach der die Studie durchgeführt wurde
Als Ergebnis dieses Schritts kennt der Hersteller die Gefährdungen des identifizierten Bestandteils anhand der vorliegenden toxikologischen Daten aus der Literatur.
Schritt 2: Risikoabschätzung
Im zweiten Schritt folgt die Risikoabschätzung. Dabei wiegt der Hersteller zwei Dinge ab:
- Die für die Kontaktart und Dauer tolerierbare Aufnahme
- Die im ungünstigsten Fall größte Exposition während der Anwendung des Produkts
Dafür sind mehrere Berechnungsschritte notwendig.
a) Berechnung der tolerierbaren Kontaktmenge oder tolerierbaren Aufnahme
Es wird die tolerierbare Menge von Kontakt bzw. Aufnahme des identifizierten Bestandteils bestimmt. Hierbei werden die tolerierbare Kontaktschwelle TCL oder die tolerierbare Aufnahme TI berechnet. Diese beschreiben die Dosis-Wirkung-Beziehung des Bestandteils, d. h. ab welcher Dosis eine toxikologische Wirkung möglich ist.
Grundlage für eine korrekte Festlegung von TCL/TI bilden die gesammelten toxikologischen Daten. Sie liefern die Information, bis zu welchem Expositionswert der Kontakt oder die Aufnahme des Bestandteils als sicher bewertet wird.
Diese Informationen werden als Point of Departure (PoD) bezeichnet. Es kommen infrage:
- Toxikologische Grenzwerte des Bestandteils (z. B. das No Adverse Effect Level, NOAEL)
- Daten eines chemischen Surrogats (mit Begründung und Unsicherheitsfaktor)
- In-silico-Daten (z. B. Verwendung von TTC-Werten der Cramer Classes)
Bei der Festlegung von TCL/TI empfiehlt sich ein konservativer Ansatz. Dazu sollte der Hersteller zusätzliche Unsicherheitsfaktoren berücksichtigen (Modifying Factors, MF). Diese Faktoren beschreiben Unsicherheiten, die durch Übertragung von Daten mit anderen Ausgangsparametern entstehen:
| Unsicherheitsfaktor | Erläuterung, Beispiel |
| Intraspezies-Variation | Berücksichtigung der Variationen innerhalb der menschlichen Population bezüglich biologischer Aufnahme, Stoffwechsel, Gewebeverteilung, Ausscheidung und Verträglichkeit von Stoffen |
| Interspezies-Variation | Liegen Daten aus Studien mit Menschen vor oder handelt es sich um Tierstudien? Berücksichtigung der Extrapolation von Daten aus Tierstudien auf den Menschen |
| Qualität der Daten | Sind die Daten aus Prüfungen nach den entsprechenden Richtlinien und GLP (Good Laboratory Praxis) durchgeführt, so ist von einer guten Qualität der Daten auszugehen. |
| Qualität der Studie | Daten sind für einen anderen Expositionsweg verfügbar oder eine andere Expositionsdauer |
Expositionsweg und Dauer | Werden z. B. Daten aus Kurzzeit-Studien für Langzeit-Exposition verwendet, ist dies über Sicherheitsfaktoren zu berücksichtigen. |
| Grenzwerte | Eine Verwendung von Grenzwerten wie Low Adverse Effect Level (LOAEL) statt No Adverse Effect Level (NOAEL) muss berücksichtigt werden, |
Alternativ dürfen die Hersteller einen toxikologisch bedenklichen Schwellenwert (TTC) für die Substanz wählen, falls nur wenige oder keine toxikologischen Daten vorliegen. Der TTC ist ein Wert, unterhalb dessen das Risiko einer Schädigung als vernachlässigbar angesehen wird.
b) Abschätzung der Expositionsdosis
Der nächste Schritt ist die Berechnung der geschätzten Worst-Case-Expositionsdosis (EEDmax) für jeden Bestandteil. Dabei werden verschiedene Faktoren berücksichtigt:
- Bioverfügbarkeit der Substanz
- Größe und Konstruktionsmaterialien des Produkts
- Beabsichtigte Verwendung
- Expositionsweg
- Patientenpopulation
- Dauer und die Häufigkeit der Verwendung
c) Ableitung der Sicherheitsspanne (Margin of Safety, MoS)
Für die Risikoabschätzung ist ein weiterer Schritt erforderlich: die Berechnung der Sicherheitsspanne (MoS) und damit die Darstellung der Akzeptanz des toxikologischen Risikos.
Die MoS wird bestimmt, indem die geschätzte Expositionsdosis mit der tolerierbaren Kontaktdosis (TCL) oder der tolerierbaren Aufnahme (TI) verglichen wird. Ein MoS-Wert > 1 weist auf ein geringes toxikologisches Risiko hin.
Im Rahmen einer chemischen Analyse nach ISO 10993-18 wurde eine Freisetzung von 150 µg/Produkt/Tag eines Bestandteils detektiert. Es wird einmalig ein Produkt pro Anwendung verwendet (Kurzzeitanwendung < 24 h).
Nach Betrachtung toxikologischer Daten kann geschlussfolgert werden, dass der Bestandteil zu 100 % vom menschlichen Körper aufgenommen werden kann. Der EEDmax ergibt für einen erwachsenen Patienten mit einem angenommenen Körpergewicht von 60 kg eine Dosis von 2,5 µg pro Kilogramm Körpergewicht.
EEDmax = Maximale Freisetzung (150 µg) / Körpergewicht (60 kg) = 2,5 µg/kg Körpergewicht
Als TI für den Bestandteil wurde über In-silico-Methoden ein Grenzwert von 30 µg/kg bw/day abgeleitet.
Hieraus ergibt sich ein MoS von 12:
MoS = TI / EEDmax = 30 / 2,5 = 12
Bei einem MoS von 12 kann davon ausgegangen werden, dass von der Freisetzung des Bestandteils kein gesundheitliches Risiko ausgeht.
Schritt 3: Risikobewertung
Im dritten Schritt entscheidet der Hersteller über die Risikoakzeptanz anhand der Kriterien, die gemäß ISO 14971 und ISO 10993-1 vorher festgelegt wurden. Diese Kriterien bestimmen, ob das geschätzte Risiko akzeptabel ist oder ob weitere Maßnahmen erforderlich sind.
Wenn die Akzeptanzkriterien nicht erfüllt sind, beispielsweise weil die Exposition gegenüber einem Bestandteil zu hoch ist, muss der Hersteller das toxikologische Risiko weiter untersuchen und zusätzliche Maßnahmen zur Risikominderung festlegen.
Zu den Möglichkeiten der Risikominimierung zählt z. B. eine Änderung der Zweckbestimmung wie der Ausschluss von bestimmten Patientengruppen.
Als Folgeaktivität können zusätzliche Tests durchgeführt werden, deren Ergebnisse eine präzisere Evaluierung und den Ausschluss der Risiken ermöglichen.
3. Praxistipps: Worauf Sie im TRA achten sollten
Tipp 1: Alle Daten einbeziehen
Beziehen Sie alle Daten aus der klinischen Anwendung in die Analyse ein. Dazu zählen:
- Anzahl und Dimension der Produkte pro Anwendung
- Patientengruppe
- Kontaktdauer (kumuliert, falls zutreffend)
- Kontaktart
Diese Punkte sind wichtig, um die Risiken durch freigesetzte Bestandteile bei der Anwendung des Produkts realitätsnah bewerten zu können und die Endpunkte gemäß ISO 10993-1 zu adressieren.
Tipp 2: Sinnvolle chemische Charakterisierung
Wählen Sie eine sinnvolle Analytik und Parameter bei der chemischen Charakterisierung, da diese meist den Hauptinput für die toxikologische Bewertung liefert.
Die Herleitung der gewählten Methode sollte sich dabei an der klinischen Anwendung des Produkts orientieren und zugleich die Anforderungen der ISO 10993-18 erfüllen. So vermeiden Sie zusätzlichen Aufwand durch die Bewertung von Bestandteilen, die in der tatsächlichen Anwendung nicht freigesetzt werden würden.
Tipp 3: Auf die Qualität toxikologischer Daten achten
Achten Sie auf die Qualität der verwendeten toxikologische Daten und dokumentieren Sie diese. So werden Tierstudien nach toxikologischen Richtlinien wie OECD und Qualitätsmanagementsystem wie GLP durchgeführt. Daten aus solchen Studien können als qualitativ angemessen eingestuft werden und eignen sich für die Herleitung von Grenzwerten.
Tipp 4: Klinische Anwendung betrachten
Achten Sie beim Ableiten von Grenzwerten unbedingt auf die klinische Anwendung Ihres Produkts und beziehen Sie diese mit ein. Sie sollten insbesondere die Expositionswege kennen, weil diese einen Einfluss auf die Grenzwerte haben.
Tipp 5: Konservativ abschätzen
Verwenden Sie immer einen konservativen Ansatz, um die Patientensicherheit zu gewährleisten.
- Liegen keine Daten zur kinetischen Freisetzung der Substanz bei längerer oder wiederholter Anwendung vor, muss man von einer kontinuierlichen Freisetzung der initialen Menge pro Tag ausgehen.
- Gehen Sie von 100 % für die Berechnung des EEDmax aus, wenn in der Literatur keine eindeutigen Daten zur Bioverfügbarkeit der Substanz vorliegen.
- Berücksichtigen Sie immer die größtmögliche Anzahl/Kontaktfläche des Medizinprodukts während der vorgesehenen Anwendung.
- Verwenden Sie für das Ableiten des TI immer den niedrigsten POD und ggf. den kritischeren Endpunkt, z. B. für chronische Toxizität bei Kurzzeitanwendung.
- Rechnen Sie mit einer geringeren Körpermasse, falls das Produkt auch bei Patienten mit geringerem Gewicht (z. B. Krebspatienten) zum Einsatz kommen kann.
Tipp 6: Experten einbeziehen
Beziehen Sie bereits bei der Planung der Biokompatibilitätsprüfungen Expert:innen ein und lassen Sie von diesen die toxikologische Risikobewertung durchführen. Profitieren Sie bei der Einschätzung der toxikologischen Daten von deren Erfahrungswissen. Mangelnde Erfahrung der bewertenden Personen kann zu Auditabweichungen führen, da es sich dabei um eine Anforderung aus der Norm handelt.
4. Die neue ISO 10993-17:2023
a) Die Änderungen
Eine Aktualisierung der ISO 10993-17, die seit über 20 Jahren in Kraft war, wurde im Herbst 2023 veröffentlicht. Damit hat die älteste Norm der ISO-10993-Familie, die von 2002 bis 2023 gültig war, eine bedeutende Revision erfahren.
Die Norm befasst sich nun nicht mehr nur mit der Festlegung von zulässigen Grenzwerten von Substanzen, sondern beschreibt auch die Durchführung der toxikologischen Bewertung eines Medizinproduktbestandteils. Entsprechend wurde der Titel der Norm angepasst. Der Umfang von 65 Seiten erklärt sich u. a. auch dadurch, dass die neue Version zahlreiche Beispiele beinhaltet.
Weitere Neuerungen sind:
- Umfangreiche Beschreibung des Prozesses mit Rechenbeispielen für Bestimmung der tolerierbaren Aufnahmemenge (TI), der tolerierbaren Kontaktmenge (TCL) und der Worst-Case-Expositionsabschätzung bezogen auf das Körpergewicht (EEDmax)
- Implementierung von Ansätzen zur Reduktion des Beurteilungsaufwands: TSL (Toxicological Screening Limit) und TQmax (freigesetzte Gesamtmenge)
- Auswahl des Ausgangswerts POD (Point of Departure) und Verwendung von Unsicherheitsfaktoren mit Angabe von “default”-Werten
- Implementierung des Margin of Safety (MoS) als für den Endpunkt spezifische Berechnungsgröße
- Erklärungen zum Dokumentationsaufwand (z. B. Begründungen für Auswahl des POD oder von Unsicherheitsfaktoren)
- Entfernung des Tolerable Exposure (TE)-Konzepts. Die klinische Anwendung wird inzwischen über die Expositionsabschätzung einbezogen.
b) Die Konsequenzen
Gute Nachricht: Toxikologische Risikobewertungen, die vor dem Inkrafttreten der überarbeiteten Norm für auf dem Markt befindliche Medizinprodukte erstellt wurden, müssen nicht an die neuen Anforderungen angepasst werden.
Trotzdem: Prüfen Sie Ihre Dokumentation hinsichtlich Aktualität und Vollständigkeit. Toxikologische Daten von Substanzen müssen aktuell sein. Ggf. gibt es Änderungen bei Materialien, Lieferanten oder beim Produktionsprozess, neue Daten oder relevante Rückmeldungen aus dem Markt, die bewertet werden müssen.
Für neue toxikologische Risikobewertungen ergibt sich durch die Aktualisierung der Norm ein deutlich höherer Dokumentationsaufwand. Dieser kann bei strukturiertem Vorgehen überschaubar gehalten werden. Beachten Sie dazu die Kapitel 2 und 3 dieses Artikels und holen Sie sich ggf. Unterstützung (siehe unten).
c) Auch die FDA spielt mit
Die FDA führt die neue Ausgabe der ISO 10993-17 als „Recognized Standard“.
5. Zusammenfassung und Fazit
Die aktuelle Ausgabe der ISO 10993-17 führt weitreichende Änderungen ein. Insbesondere beschreibt sie einen Prozess zur toxikologischen Risikoabschätzung und unterstützt die Leser mit vielen Beispielen.
Die Norm legt fest, dass der Prozess der toxikologischen Risikobewertung aus insgesamt drei Schritten besteht:
- Identifikation von Gefährdungen
- Risikoabschätzung
- Risikobewertung
Wie die Hersteller diese drei Schritte am besten durchlaufen, beschreibt dieser Artikel.
Hersteller, die die neuste Version der Norm noch nicht berücksichtigen, sollten ihre Dokumentation hinsichtlich Aktualität und Vollständigkeit prüfen (s. Kapitel 4.b).
Falls auch Sie vor der Aufgabe stehen, die Biokompatibilität Ihres Produkts beurteilen zu müssen, dann hilft Ihnen das Team des Johner Instituts bei allen Aktivitäten.
- Wir führen die komplette toxikologische Risikobewertung durch oder unterstützen Sie dabei.
- Wir helfen Ihnen bei der Gap-Analyse, um sicherzustellen, dass die Bewertung der toxikologischen Risiken dem Stand der Technik entspricht und Ihre Audits sowie Zulassungsverfahren reibungslos verlaufen.
- Wir planen, organisieren und bewerten Ihre Biokompatibilitätsprüfungen von Medizinprodukten, u. a. durch chemische Analysen zur Materialcharakterisierung.
Melden Sie sich hier, wenn Sie Unterstützung wünschen. Wir sind für Sie da!
Änderungshistorie
- 2025-12-16: Anpassung des Zitats aus der ISO 10993-1 entsprechend der neuesten Normenversion (2025)
- 2024-03-02: Hinweis auf die Anerkennung als Recognized Standard durch die FDA in Kapitel 4.c) ergänzt
- 2024-02-13: Erste Version des Artikels erstellt


