
Für Hersteller ist die Antwort auf die Frage relevant, ob und wann beim Einsatz künstlicher Intelligenz in Medizinprodukten klinische Studien notwendig sind. Denn davon hängen die Dauer und die Kosten ab, um diese Produkte in den Markt zu bringen.
Die gute Nachricht vorweg: Es gibt Fälle, in denen die Hersteller auf klinische Studien bei Produkten mit KI verzichten können. Welche Fälle das sind, klärt dieser Artikel.
1. Zusammenfassung für die eiligen Leser
In den meisten Fällen wird es den Herstellern nicht gelingen, anhand von veröffentlichten klinischen Daten die Konformität mit den regulatorischen Anforderungen (z. B. an die Leistungsfähigkeit) nachzuweisen. Das liegt daran, dass sich die Äquivalenz der Produkte bzw. KI-Modelle, mit denen die Daten gewonnen wurden, nicht ausreichend darlegen lässt.
Allerdings besteht die Möglichkeit, durch retrospektive Analysen auf prospektive klinische Studien bei Produkten mit KI zu verzichten.
2. Die Rechtslage
Die Rechtslagen in der EU und den USA unterscheiden sich. Die FDA definiert in ihrem Guidance-Dokument über Clinical Decision Support Software vier Kriterien, die eine Software-Anwendung erfüllen muss, um in den USA nicht als Medizinprodukt zu gelten und nicht von der FDA überwacht zu werden. Diese Voraussetzungen werden jedoch von KI-basierter Software meist nicht erfüllt, beispielsweise weil die Software der Analyse von Bildern oder Signalen dient.
Lesen Sie hier mehr zu Qualifizierung von Software als Medizinprodukt und dazu, wie die FDA (Clinical) Decision Support Systems einschätzt.
Weiterhin gibt es Beiträge zu den regulatorischen Anforderungen an KI-basierte Medizinprodukte und zu den Anwendungen der KI in der Medizin.
Die MDR (analog die IVDR) verpflichtet die Hersteller, die Sicherheit, Leistung und den klinischen Nutzen der Produkte im Rahmen der klinischen Bewertung bzw. der Leistungsbewertung von IVD nachzuweisen. So unterschiedlich die Anforderungen von MDR und IVDR an den klinischen Nachweis für Medizinprodukte bzw. IVD sein mögen, so vergleichbar sind sie für Software. Die Besonderheiten und Gemeinsamkeiten bei der klinischen Bewertung bzw. Leistungsbewertung von Medizinprodukte- bzw. IVD-Software beschreibt die MDCG in ihrem lesenswerten Guidance-Dokument Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software von 2020.
Falls nicht ausreichend klinischen Daten vorliegen, muss der Hersteller diese generieren. In der Regel bedeutet dies, dass eine prospektive klinische Studie durchgeführt werden muss.
Es ist (nahezu) ausgeschlossen, dass man für eine KI-basierte Software die klinische Leistung anhand von Literaturdaten aufzeigen kann. Das gelingt fast nur, wenn man die Publikation selbst geschrieben und somit die Studie durchgeführt hat und die Rohdaten vorliegen. In diesem Fall ist es meist einfacher, direkt die Studiendaten zu verwenden und sich den Umweg über die Publikation zu sparen.
Dennoch ist es strategisch häufig sinnvoll und möglich, auf bereits vorliegende Datensätze zurückzugreifen – vorausgesetzt, dass die Datensätze nicht zum Trainieren und Testen eingesetzt wurden und es sich um einen unabhängigen Validierungsdatensatz mit entsprechender statistisch relevanter Fallzahl für alle Subpopulationen handelt.
Die MDR spricht von klinischen Prüfungen und die IVDR von klinischen Leistungsstudien, jedoch nicht von klinischen Studien. Dieser Artikel verwendet beide Begriffe synonym.
3. Das Problem mit der Äquivalenz
a) Anforderung der MDR
Insbesondere unter der MDD war es üblich, klinische Daten von bereits bestehenden Produkten zu verwenden und damit auf klinische Studien zu verzichten. Voraussetzung war, dass die Daten von äquivalenten Produkten stammen. Dazu zählt auch deren technische Äquivalenz. Der erforderliche Nachweis der technischen Äquivalenz betrifft explizit auch die Software-Algorithmen:
Technisch: Das Produkt ist von ähnlicher Auslegung, wird unter ähnlichen Anwendungsbedingungen angewandt, hat ähnliche Spezifikationen und Eigenschaften einschließlich […] Software-Algorithmen […] und hat ähnliche Funktionsgrundsätze und entscheidende Leistungsanforderungen.
MDR, Anhang XIV, 3. Absatz, 1. Spiegelstrich
Bei Medizinprodukten der Klasse III und bei implantierbaren Produkten wird es noch schwieriger, auf klinische Studien zu verzichten:
[…] CLINICAL EVALUATION of class III and implantable devices (MDR) shall include data from a CLINICAL INVESTIGATION unless the conditions of Article 61(4), (5) or (6) of the MDR have been fulfilled.
MDCG 2020-1, Kapitel 4.4
b) Technische Äquivalenz bei künstlicher Intelligenz
Bei Medizinprodukten, die Verfahren der künstlichen Intelligenz und dabei insbesondere des Machine Learnings verwenden, entsprechen die Modelle den Software-Algorithmen (z. B. Modelle wie die verschiedenen Ausprägungen neuronaler Netzwerke). Diese unterscheiden sich bezüglich
- Architektur der Modelle, z. B.
- Anzahl der Layer
- Anzahl der Neuronen pro Layer
- Verknüpfung der Layer
- Aktivierungsfunktionen (z. B. RELU)
- Verknüpfung der Teilmodelle
wie bei LSTM oder LLM - Werte der gefitteten Parameter
(Gewicht und Bias)
Die Hyperparameter zum Trainieren der Modelle wirken sich auf die gefitteten Parameter aus. Ihre Wahl ist aber kein Aspekt der technischen Äquivalenz. Hingegen zählen die „Anwendungsbedingungen“ zur technischen Äquivalenz.
Bisher gibt es keine Vorgaben, was die Anwendungsbedingungen in diesem Kontext umfassen. Mögliche Aspekte sind:
- Technische Umgebung wie Hardware und Betriebssystem
- Programmiersprache und verwendete Bibliotheken
- Verwendung und Einbettung des Modells in das gesamte Medizinprodukt
c) Nachweis der technischen Äquivalenz
In der Regel wird es Herstellern nicht gelingen, die Äquivalenz der „Software-Algorithmen“ nachzuweisen, ohne den Software-Code des Äquivalenzprodukts zu vergleichen. Das ist, wie die MDCG feststellt, auch nicht notwendig.
„It is the functional principle of the software algorithm, as well as the clinical performance(s) and intended purpose(s) of the software algorithm, that shall be considered when demonstrating the equivalence of a software algorithm. It is not reasonable to demand that equivalence is demonstrated for the software code, provided it has been developed in line with international standards for safe design and validation (IEC 62304, and IEC 82304-1) of medical device software.“
Kommentar in MDCG 2020-5, Kapitel 3.1 (b)
Bei KI-Modellen ist die Äquivalenz nicht einmal bei identischem Software-Code nachgewiesen. Denn der Software-Code (als Teil des Medizinprodukts) definiert das KI-Modell, aber nicht die Werte der gefitteten Parameter. Diese sind bestimmt durch die Trainingsdaten und die Art des Trainings (z. B. Hyperparameter). Der Software-Code für das Training ist jedoch kein Teil des Medizinprodukts und wird von der MDCG 2020-5 nicht abgedeckt.
Die MDCG 2020-5 verlangt, das „functional principle“ und die „clinical performance“ (dazu später mehr) beim Äquivalenzvergleich zu verwenden. Bei KI-Anwendungen ist es die Modellarchitektur, die am ehestem dem „functional principle“ entspricht.
Es gibt derzeit keine einheitliche Einschätzung der Behörden und Benannten Stellen, ob eine gleiche oder ähnliche Modellarchitektur ausreicht, um die technische Äquivalenz nachzuweisen. Daher sollten Hersteller immer die „clinical performance“ beider Produkte bestimmen und vergleichen. Diese Vergleiche müssen auf angemessenen wissenschaftlichen Begründungen beruhen. Ohne Zugriff auf das andere Modell und/oder die Technische Dokumentation des anderen Produkts wird ein solcher Vergleich nur schwer zu führen sein.
Falls es eine CE-markierte KI-Software mit derselben Zweckbestimmung auf dem Markt gibt, dann wäre eben jener Vergleich der klinischen Leistung der beiden MDSW-Produkte das Ziel der benötigten klinischen Studie.
Die bereits zugelassene KI mit derselben Zweckbestimmung wäre die Vergleichsmethode, und es würden die verfügbaren Validierungsdatensätze mit beiden KI-Software-Produkten analysiert und der Anteil der Übereinstimmung (percent of agreement) untersucht werden. Ggf. würde man eine weitere Referenzmethode hinzuziehen – je nach Güte der bereits zugelassenen KI-Software und der klinischen Strategie bzw. Leistungsbewertungsstrategie.
Es bedarf ausreichend klinischer Daten, um die Konformität mit den grundlegenden Sicherheits- und Leistungsanforderungen nachzuweisen. Klinische Daten lassen sich auch auf anderen Wegen als durch klinische Studien sammeln (s. nächstes Kapitel).
4. Nachweis der Konformität
a) Nachzuweisende Aspekte/Anforderungen
Mit klinischen Daten müssen die Hersteller die Sicherheit, Leistungsfähigkeit und den klinischen Nutzen ihrer Medizinprodukte bzw. IVD nachweisen. Im Fall von „Medical Device Software“ (MDSW) bedeutet dies gemäß MDCG 2020-1 den Nachweis von:
- Clinical association / scientific validity
- Technical performance / analytical performance
- Clinical performance
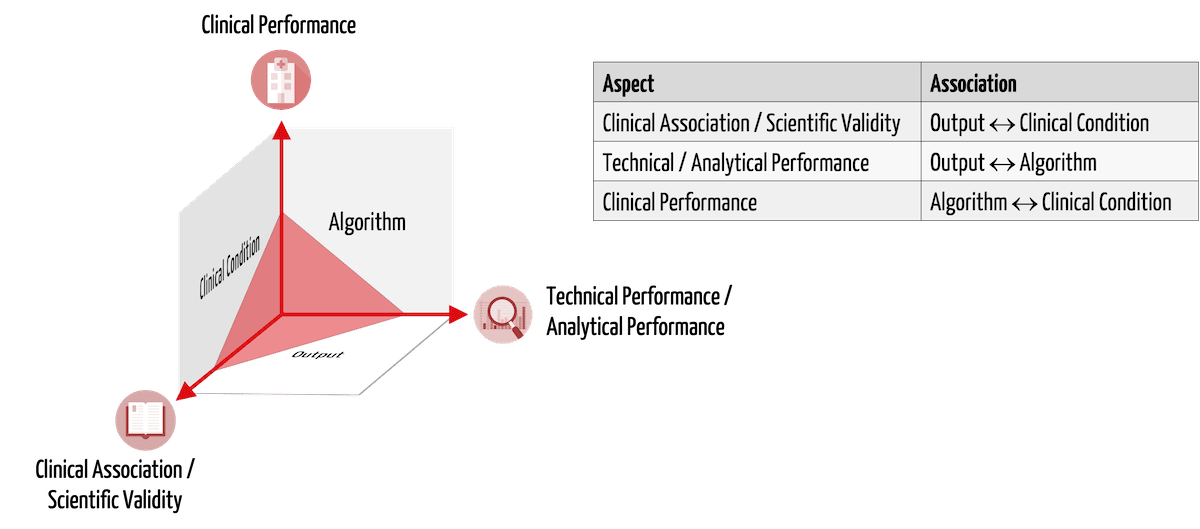
MDSW umfasst Standalone-Software und Software als Teil eines Medizinprodukts bzw. IVDs, wenn es direkt dessen Zweckbestimmung dient. Ein Beispiel dafür ist die Software eines Defibrillators, die den Herzschlag eines Patienten erkennt.
Tabelle 1 stellt diese drei Aspekte des klinischen Nachweises am Beispiel einer KI-basierten Medizinprodukte-Software vor, welche in CT-Bildern krebsverdächtiges Gewebe identifiziert.
| Nachzuweisender Aspekt | Beschreibung | Beispiel |
| Clinical association / scientific validity | Es gibt einen Zusammenhang zwischen dem Output der Software und dem zu prüfenden physiologischen/klinischen Zustand. | Die Helligkeitswerte1), die die Software markiert, beschreiben tatsächlich Krebsgewebe. |
| Technical performance / analytical performance | Gegebene Inputs führen unter Verwendung des Algorithmus reproduzierbar zu korrekten Outputs. | Die Software markiert in den Bildern korrekt und reproduzierbar die Stellen mit bestimmten Helligkeitswerten1. |
| Clinical performance | Die Zweckbestimmung wird für alle vorgesehenen Patienten (mit jeweiligen Krankheiten) im entsprechenden Nutzungskontext durch die vorgesehenen Nutzer erreicht. | Die vorgesehenen Anwender erkennen den Krebs auch in Bildern von anderen CTs, bei Patienten in Bauchlage, bei gleichzeitigen Verkalkungen des Gewebes usw. korrekt. Metriken wie die diagnostische Sensitivität und Spezifität bestimmen die klinische Leistungsfähigkeit. |
1) Bei CTs entsprechen die Helligkeitswerte den Hounsfield-Units (HU), manchmal auch als „Röntgendichte“ bezeichnet.
b) Methoden zum Nachweis
Die Leitlinie MDCG 2020-1 schlägt verschiedene Methoden vor, um die o.g. Aspekte nachzuweisen.
| Nachzuweisender Aspekt | Methoden (Beispiele) |
| Clinical association / scientific validity | Systematische Literaturrecherche in medizinischen Leitlinien und wissenschaftlicher Literatur (Es ist z. B. beschrieben, welche „Helligkeitswerte“ Krebsgewebe hat.) Klinische Prüfungen bzw. klinische Leistungsstudien für IVD-/MD-Software |
| Technical performance / analytical performance | Software-Testing auf Systemebene zum Nachweis der folgenden Leistungsparameter: analytische Sensitivität, analytische Spezifität, Richtigkeit (Verzerrung), Präzision (Wiederholbarkeit und Reproduzierbarkeit), Genauigkeit (als Ergebnis von Richtigkeit und Präzision), Nachweis- und Quantifizierungsgrenzen, Messbereich, Linearität, Cut-off. (Diese Tests werden ergänzend durchgeführt zu bzw. basierend auf den Unit- und Integration-Tests gemäß IEC 62304, dem Nachweis der IT-Sicherheit und der Gebrauchstauglichkeit; vgl. die Kriterien der ISO 25010.) |
| Clinical performance | Retrospektive Analysen vorhandener Daten in ausreichender Qualität und Quantität Klinische Prüfungen (bzw. klinische Leistungsstudien für IVD-/MD-Software) Simulationen Klinische Literatur (setzt Äquivalenz – auch die klinische Äquivalenz – voraus) kann ggf. als Basis oder ergänzend genutzt werden. |
Die Validierung der KI-Software gegen die Zweckbestimmung umfasst neben dem Nachweis der klinischen Leistung auch Usability-Tests (summative Evaluation) zum Nachweis der Gebrauchstauglichkeit.
Das GHTF-Dokument Software as a Medical Device: Clinical Evaluation nennt weitere Methoden und erläutert die nachzuweisenden Aspekte.
5. Sammeln klinischer Daten
Der Nachweis der klinischen Leistungsfähigkeit („clinical performance“) stellt für Hersteller KI-basierter Medizinprodukte die größte Hürde dar. Das bedeutet aber nicht, dass prospektive klinische Studien immer notwendig sind.
a) Retrospektive Analysen
Bei einer retrospektiven Analyse nutzen Hersteller bereits vorhandene Daten.
Im Fall einer Software zur Krebserkennung könnte der Hersteller vorhandene CT-Aufnahmen nutzen, diese durch die Software bewerten lassen und mit vorhandenen Krebsdiagnosen vergleichen. Dies funktioniert aber nur, wenn folgende Voraussetzungen erfüllt sind:
- Die Bilder wurden unter vergleichbaren technischen Rahmenbedingungen gewonnen, wie sie vom bestimmungsgemäßen Gebrauch vorgesehen sind. Dazu zählen die Aufnahmegeräte (CTs), Aufnahme-Parameter (z. B. Strahlungsintensität, Winkel, Patientenpositionierung, mit/ohne Kontrastmittel) und Bildparameter (Größe und Auflösung der Bilder, Kodierung der Hounsfield-Werte)
- Das Patientenkollektiv, mit dem die Bilder aufgezeichnet wurden, ist vergleichbar mit dem in der Zweckbestimmung vorgesehenen Kollektiv. Dazu zählen anatomische und morphologische Eigenschaften ebenso wie verschiedene Orte, Größen und Arten des Tumors und möglicher Ergüsse.
- Die Fallzahl ist ausreichend und die Qualität der Daten ist wissenschaftlich valide.
- Die Einverständniserklärung zur Nutzung der Daten für klinische Studienzwecke liegt vor.
- Auch die Vergleichbarkeit der Nutzer und der Nutzungsumgebung müssten diskutiert werden. Allerdings lassen sich Usability-Tests leichter nachholen als klinische Studien.
b) Klinische Studien bei künstlicher Intelligenz
Falls diese Voraussetzungen nicht oder nicht vollständig erfüllt sind, müssen Hersteller die klinischen Daten generieren, typischerweise durch eine prospektive klinische Prüfung (bzw. klinische Leistungsstudie bei IVD-/MD-Software).
Das wäre beispielsweise notwendig, falls die Software auch für Patienten mit einer Krebserkrankung genutzt werden soll, zu denen noch keine ausreichenden klinischen Daten vorliegen. Im konkreten Fall müsste die klinische Studie zumindest die korrekte Segmentierung des Krebsgewebes für dieses bislang nicht überprüfte Patientenkollektiv nachweisen.
6. Fazit
In der Regel scheitert der Versuch, die Äquivalenz von KI-Modellen zu beweisen und die klinische Leistungsfähigkeit der Software mittels Literatur zu belegen. Viele Hersteller werben sogar explizit mit „besseren“ Modellen.
Das bedeutet aber nicht, dass bei KI-basierten Medizinprodukten zwingend eine prospektive klinische Studie notwendig wird. Oft ist es möglich, diese Studien durch retrospektive Analysen bereits verfügbarer Daten ausreichender Qualität und Quantität ganz oder teilweise zu vermeiden.
Durch eine klug gewählte klinische Strategie bzw. Leistungsbewertungsstrategie und eine fokussierten Zweckbestimmung gelingt es, das Produkt schneller in den Markt zu bringen. Während der anschließenden Anwendung des Produkts werden klinische Daten erzeugt, die Sie z. B. als PMCF- bzw. PMPF-Studien auswerten und für eine Erweiterung der Zweckbestimmung nutzen können.
Melden Sie sich, um mit den klinischen Expertinnen und Experten des Johner Instituts die beste klinische Strategie bzw. Leistungsbewertungsstrategie für Ihr Produkt zu erarbeiten. Sie sparen dadurch Zeit und Geld (z. B. für klinische Studien bei künstlicher Intelligenz) und können Ihr Produkt schnell und konform in den Verkehr bringen.


Vielen Dank für den informativen Fachartikel.
Eine Frage noch: gemäß BfArM Leitfaden zur regulatorischen Einordnung klinischer Studien mit Medizinprodukten würde es sich bei der Retrospektive Analyse in 5 b) auch um eine klinische Prüfung handeln, wenn diese an (in Bezug auf eine Person) identifizierbaren menschlichen Daten wie z.B. Bilddaten durchgeführt werden (Fallbeispiel A.9 aus dem Leitfaden).
Also wäre eine weitere Voraussetzung, dass die Bilddaten anonymisiert sind um nicht eine klinische Prüfung anmelden zu müssen? Oder kommen Sie da zu einer anderen Einschätzung?
MfG
Anne Schmidt
Sehr geehrte Frau Schmidt,
besten Dank für Ihre Nachricht und Ihre spannende Frage!
Sie beziehen sich in diesem Fall vermutlich auf den Absatz der unter dem Fallbeispiel A9 integriert ist und auf die Deklaration von Helsinki verweist.
In diesem Abschnitt wird darauf hingewiesen, dass eine systematische Retrospektive Befundung und somit die Forschung an identifizierbaren menschlichen Materialien und Daten, eine klinische Studie darstellt. Beachten Sie, dass nicht jede „klinische Studie“ auch eine „klinische Prüfung“ ist.
In diesem Fall müssen wir uns zunächst die Definitionen der Begriffe „klinische Studie“ und „klinische Prüfung“ anschauen. Als klinische Studie werden in dem Leitfaden unter Kapitel 3 alle „systematischen Untersuchungen mit mindestens einem menschlichen Prüfungsteilnehmer“ bezeichnet, im Gegenzug dazu sind „klinische Prüfungen“ unter Artikel 2, Nr. 45 der MDR definiert als „„klinische Prüfung“ bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird“.
Diese „klinischen Prüfungen“ werden weiter in unterschiedliche Arten der Prüfung, je nach Zielsetzung, unterteilt. Mehr Informationen zu dem Thema der unterschiedlichen klinischen Prüfungen nach MDR finden sie in unserem Blogartikel „Klinische Prüfungen von Medizinprodukten im Rahmen der MDR – Der regulatorische Weg“ (https://www.johner-institut.de/blog/regulatory-affairs/klinische-pruefungen-von-medizinprodukten/)
Der Hinweis unter dem Fallbeispiel A9 zielt darauf ab, dass jegliche „klinischen Studie“, sei es einer „klinischen Prüfung“ oder einer „klinischen Studie“ nach z.B. Berufsordnung für Ärzte (BoÄ), an bestimmte Voraussetzungen bezüglich des Datenschutzes gebunden ist, um die Identität der Teilnehmer zu schützen. Dazu müssen die Daten nicht zwingend anonymisiert werden.
Die Nutzung von identifizierbaren menschlichen Materialien oder Daten geht nicht zwingend mit der Durchführung einer „klinischen Prüfung“ einher.
Viele Grüße,
Susanne Golombek