
Mit zunehmender Vernetzung von Medizinprodukten stehen Sie immer häufiger vor der Frage, was das Medizinprodukt ist und ob Sie besser das „ganze System“ d.h. die Kombination von Medizinprodukten oder lieber die Komponenten einzeln als Medizinprodukt in Verkehr bringen sollen.
Ein Beispiel für die Kombination von Medizinprodukten
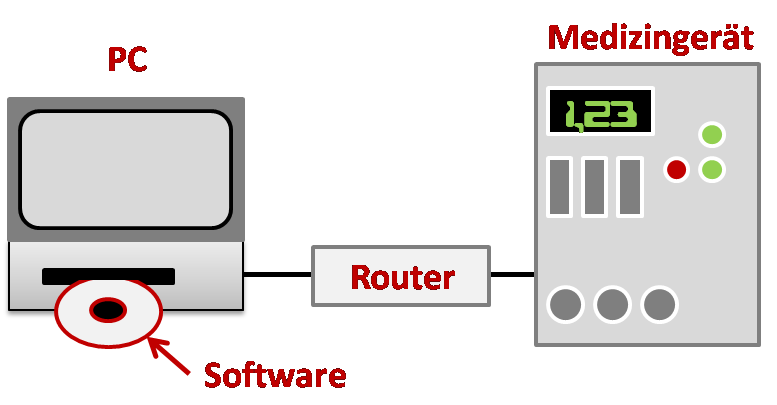
In diesem Beispiel besteht das ganze System aus einer stand-alone Software, einem PC, auf dem die Software läuft, aus einem Router und weiteren Netzwerkkomponenten und aus einem klassischen Medizingerät, dessen Daten auf dem PC angezeigt werden bzw. das von diesem gesteuert wird.
Die Antwort auf die Frage, was denn hier das Medizinprodukt sei, ist ebenso schnell wie einfach beantwortet: Das, was der Hersteller als solches in Verkehr bringt, um Menschen zu diagnostizieren und zu behandeln, ist das Medizinprodukt. Wenn der Hersteller sagt, die Software alleine sei ein Produkt, mit dem Menschen diagnostiziert oder behandelt werden, und diese einzeln verkauft, dann ist diese Software das Medizinprodukt. Wenn der Hersteller die Software zusammen mit dem PC in Verkehr bringt, dann ist diese Kombination das Medizinprodukt. Der Hersteller könnte sogar die Gesamtheit, d.h. die Software, den PC, den Router und das Gerät als ein Medizinprodukt in Verkehr bringen.
Kombination von Medizinprodukten: Rechtliche Grundlage
Der §10 des Medizinproduktegesetzes sagt:
(1) Medizinprodukte, die eine CE-Kennzeichnung tragen und die entsprechend ihrer Zweckbestimmung innerhalb der vom Hersteller vorgesehenen Anwendungsbeschränkungen zusammengesetzt werden, um in Form eines Systems oder einer Behandlungseinheit erstmalig in den Verkehr gebracht zu werden, müssen keinem Konformitätsbewertungsverfahren unterzogen werden. Wer für die Zusammensetzung des Systems oder der Behandlungseinheit verantwortlich ist, muss in diesem Fall eine Erklärung nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1 abgeben.
(2) Enthalten das System oder die Behandlungseinheit Medizinprodukte oder sonstige Produkte, die keine CE-Kennzeichnung nach Maßgabe dieses Gesetzes tragen, oder ist die gewählte Kombination von Medizinprodukten nicht mit deren ursprünglicher Zweckbestimmung vereinbar, muss das System oder die Behandlungseinheit einem Konformitätsbewertungsverfahren nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1 unterzogen werden.
Das bedeutet für Hersteller, die für die Software und das Medizingerät das Konformitätsbewertungsverfahren jeweils bereits durchlaufen haben: Die Kombination aller Produkte, die einen handelsüblichen Router enthält, muss eine erneute Konformitätsprüfung durchlaufen.
Sie finden hier eine ausführliche Darstellung der rechtlichen Anforderungen an Systeme und Behandlungseinheiten.
Abwägungen bei der Kombination von Medizinprodukten
Ich halte eine andere Frage für die spannendere: Was sollte der Hersteller als Medizinprodukt in Verkehr bringen? Hier ist meine Antwort etwas differenzierter, denn es gibt Vor- und Nachteile der verschiedenen Ansätze.
Gründe gegen die Kombination von Medizinprodukten zu einem Produkt
Für eine Inverkehrbringung von möglichst granularen Produkten (hier Software und Medizingerät einzeln) sprechen die folgenden Gründe:
- Flexibles Kombinieren
Sie sind freier in der Kombination: Solange die einzelnen Medizinprodukte ein CE-Zeichen haben und in der ursprünglichen Zweckbestimmung eingesetzt werden, können Sie alle Kombinationen vermarkten, die nur beide Produkte enthalten. Ohne eine erneute Konformitätsprüfung. - Einfacherer Austausch
Sie sind nicht so abhängig von Komponenten wie dem Router: Gerade Netzwerkkomponenten sind einem schnellen Technologiewandel unterworfen, und Sie wollen nicht bei jeder neuen Generation die Konformitätsprüfung erneut durchlaufen. - Konformitätsbewertung / Zulassung
Wenn es Änderungen an einem der Medizinprodukte gibt, z.B. bei der Software, so müssen Sie nur die Software, nicht aber die vollständige Kombination neu „zulassen“.
Gründe für die Kombination von Medizinprodukten zu einem Produkt
Es gibt allerdings auch Gründe, welche gegen eine Inverkehrbringung der einzelnen Medizinprodukte sprechen:
- Ökonomische Erwägungen
Wenn Sie die Software, um ein Beispiel zu nennen, ohne den PC in Verkehr bringen, werden Ihre Kunden preiswerte Hardware einkaufen. Ein Medizinprodukt, welches die gleiche Hardware und Software kombiniert, ließe sich wahrscheinlich mit einer höheren Marge vertreiben. - Risikobeherrschung
Wenn Sie alle Komponenten, in unserem Beispiel inklusive dem Router, in Verkehr bringen, so haben Sie auch die Kontrolle über alle Komponenten. Dies kann bei der Risikoanalyse und Risikokontrolle einiges vereinfachen. So hätten Sie die Möglichkeit, bei einem Router (den Sie herstellen) gewisse Risiken, welche durch eine fehlerhafte Datenübertragung verursacht werden, durch eigene Prüfroutinen zu erkennen und geeignet zu reduzieren.
Kombinationen von Produkten beim Betreiber
Bei der Kombination von Medizinprodukten muss unterschieden werden, ob der Hersteller diese Kombination in Verkehr bringt oder ob der Betreiber diese Produkte erst bei sich kombiniert. Die Hersteller müssen auch den zweiten Fall im Blick haben.
Beispielsweise fordert die MDR, dass die Hersteller genaue Vorgaben dazu machen, welche Voraussetzungen die Betreiber erfüllen müssen, um diese Kombinationen sicher zu ermöglichen. Diese Vorgaben müssen z.B. die IT-Umgebung (Netzwerkbandbreiten, Verantwortlichkeit für Virenschutz etc.) umfassen.
Fazit: Die Entscheidung, was nun das Medizinprodukt ist, ist weit mehr als eine regulatorische Angelegenheit. Hier spielen Marketinggesichtspunkte und das Risikomanagement eine wichtige Rolle.



Sehr geehrte Damen und Herren,
vielen Dank für den aufschlussreichen Bericht. Mich würde eine andere Frage viel mehr interessieren: Wie soll sich eine Firma verhalten, die in ein bestehendes System (Kombination aus alten (also vor der 3. Ausgabe der EN 60601-1) zugelassenen Medizinprodukten) neue Rechner einbringen möchte. Muss ein Konformitätsbewertungsverfahren nach MPG § 10 Abs 2 durchgeführt werden ?
Vielen Dank
Sehr geehrter Herr Hempfling,
danke für die spannende Frage! Wenn Sie einen Rechner in einem Medizinprodukt austauschen, dann ist eine neue Konformitätsbewertung notwendig. Schließlich könnte – um im IEC 60601-Kontext zu bleiben — nicht sichergestellt, dass das Produkt mit dem neuen Rechner beispielsweise noch immer die EMV-Anforderungen erfüllt.
Der §10 meint nicht Komponenten, sondern spricht eher von Systemen, die aus Produkten kombiniert werden.
Viele Grüße, Christian Johner
Sehr geehrter Professor Dr. Johner,
aus gelesenem stellt sich bei mir die Frage was unterscheidet Kombinationen von einem System?
Auch was ist unter erstmaligen in Verkehr bringen zu verstehen.
Fall 1
Der Zusammensetzer des Hersteller baut ein System mit neuen eigenen Produkten beim Kunden auf einen neuen mobilen medizinischen Gerätewagen (MP).
Fall 2
Der Zusammensetzer des Hersteller baut ein System mit neuen eigenen Produkten beim Kunden auf einen neuen mobilen medizinischen Gerätewagen aus dem Bestand des Kunden.
Vielen Dank und Grüße
Sehr geehrter Herr Liebscher,
danke für die spannende Frage! Aus der Beschreibung der Fälle kann ich die Antwort nicht geben. So wie Sie das schildern, könnte es sein, dass gar keine Inverkehrbringung vorliegt, sondern eine Eigenherstellung. Wenn Sie im Auftrag des Kunden den Gerätewagen aus Produkten (gleich ob Neu- oder Altprodukten) zusammenbauen, wäre das eine Eigenherstellung. Bei dem Wagen mit dem Altprodukt drängt sich dieser Verdacht besonders auf.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
gern würde ich eine Frage zu der Kombination von zwei einzeln zugelassenen Medizinprodukten stellen.
Wenn zwei Produkte zugelassen sind, eines dieser Geräte ist das Grundgerät, das zweite dient als Zubehör.
Sofern das Grundgerät Klasse 1 und das Zubehör Klasse2a ist, können beide mit den jeweiligen Klassen In-Verkehr gebracht werden oder muss das Produkt Klasse 1 höher gestuft werden?
Das MPG sagt hierzu, dass beide Geräte als eigenständiges Medizinprodukt zu behandeln sind. Wie ist das bei der MDR?
Danke und beste Grüße
Sehr geehrte Frau Zeiher,
Sie können weiter beide Produkte unabhängig voneinander in den Verkehr bringen und unabhängig voneinander klassifizieren. Es gibt weiter Ausnahmen wie z.B. die Regel die besagt, dass eine Software (z.B. Zubehör), die ein MP steuert und beeinflusst, in die gleiche Klasse fällt.
Dass das Zubehör und das Produkt eine andere Klasse haben, ist nicht unüblich. Das wird auch so bleiben.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
Was ist mit Produkten welche vom Design her von einer Softwareplattform abhängig sind? Typischerweise haben Softwareplattformen nicht nur eine Funktion sondern interagieren mit verschiedenen Produkten, sind aber klar Teil der Auswertung (Risiko).
Als Beispiel:
– Ein Wearable, dass bestimmte Parameter misst und die Plattform analysiert die Daten. Die Daten werden auf dem Smartphone analysiert.
– Ein Laborkit für IVD muss durch Softwarepipelines für eine komplexe Analyse. Die Daten/Auswertung werden auf eine geteilten Frontend Plattform ausgeben.
Was sind nun einzelne Produkte? Alle einzelnen Komponenten oder ein Komplettlösung (Kit +Softwareplattform in der cloud + Frontend) oder (Wearable+Softwareplattform+App)?
Vielen Dank
Freundliche Grüsse
Sehr geehrter Herr Müller,
die Definition dessen, was das Medizinprodukt bzw. was die Medizinprodukte sind, legen Sie(!) fest. D.h. Sie haben die Möglichkeit z.B. alles als ein Produkt in den Verkehr zu bringen oder als getrennte Produkte z.B. das Wearable und die cloudbasierte Software-Plattform.
Jede dieser Entscheidungen hat Pros und Cons.
Beste Grüße, Christian Johner
Sehr geehrter Prof. Johner,
ich möchte ein Messgerät (Nicht-Medizinprodukt) an der Probenahmestelle eines Medizinproduktes fest per Schlauch anschließen.
Das Messgerät wird nur außerhalb der Behandlung benutzt und ist elektrisch vom Medizinprodukt getrennt.
Ist dies aus Ihrer Sicht so möglich?
Vielen Dank vorab und Grüße
Florian Engeln
Lieber Herr Engeln,
danke für die spannende Frage! Wenn das Medizinprodukt gekennzeichnet ist, dass es nicht für Behandlungen genutzt werden kann, wäre es kein Problem. Ansonsten wäre es okay, wenn das im Rahmen der Zweckbestimmung geschehen wäre. Das ist wahrscheinlich aber nicht der Fall.
Bei der Veränderung eines bereits in den Verkehr gebrachten Produkts muss der Betreiber die Sicherheit gewährleisten und z.B. nachweisen, dass die elektrische Trennung wirksam ist und keine nachteiligen Auswirkungen der Veränderung bestehen.
Wenn das Produkt erst in den Verkehr gebracht werden soll, wäre eine Konformitätsbewertung notwendig. Das ist hoffentlich nicht der Fall.
Beste Grüße nach SW, D15, EK1 oder wo Sie gerade sind, Christian Johner
Sehr geehrter Herr Johner,
wir entwickeln aktuell ein Medizinprodukt, welches als funktionale Komponente ein zugekauftes Medizinprodukt enthalten soll. Dieses zugekaufte Medizinprodukt würde aber innerhalb unseres zu entwickelnden Produktes nicht die ursprüngliche Zweckbestimmung erfüllen.
Sehen Sie hier ein Problem bei der Zulassung ? Muss der Hersteller des zugekauften Medizinproduktes über die Verwendung informiert werden? Ich sehe hier auch eine Problematik im Bereich PMS für den Inverkehrbringer.
Sehr geehrter Herr Kaufmann,
es spricht nichts dagegen, das andere Medizinprodukt zu integrieren, auch wenn das nicht in der ursprünglichen Zweckbestimmung eingesetzt wird. Sie müssen aber gewährleisten, dass Ihr Produkt insgesamt die grundlegenden Anforderungen erfüllt.
Sie sollten den Hersteller des ursprünglichen Medizinprodukts nicht nur informieren, sondern idealweise über eine QSV dazu verpflichten z.B. gefundene Fehler und Änderungen am Produkt (vorab) Ihnen zu kommunizieren.
Beste Grüße, Christian Johner
Hallo Herr Johner,
das ist schonmal eine beruhigende Antwort, vielen Dank. Leider bietet die MDR meines Wissens nach keine Aussagen zu dieser speziellen Konstellation.
Eine letzte Frage:
Müsste von unserer Seite dann auch die komplette technische Dokumentation zu dem integrierten MP vorgehalten werden oder ein separates PMS geführt werden? Wir befürchten, dass durch die zu geringe Abnahmemenge vielleicht kein Interesse an einer QSV besteht.
Danke im Voraus !
Sehr geehrter Herr Kaufmann,
es muss die komplette TD für Ihr MP vorliegen. Wie viel vom „eingebauten“ MP vorhanden sein muss, hängt von der Systemarchitektur und dem Risiko durch die Komponente = MP ab.
Ein separates PMS ist nicht notwendig.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
Vielleicht könnten Sie mir bei folgendem Szenario helfen:
Einerseits haben wir ein MP, welches als nicht-invasives Produkt Messfunktionen durchführt. Des Weiteren wird für die tatsächliche Messung ein weiteres MP benötigt, welches aufgrund des verwendetem Zubehörs als invasives Produkt zählt. Die Messung ist ohne Kombination beider Geräte nicht möglich, jedoch werden beide MP separat in Verkehr gebracht. Meine daraus resultierende Frage lautet: Zählt MP #1 ebenfalls als invasiv, da die Messungen nicht ohne dem invasiven MP #2 durchgeführt werden kann oder wird MP #1 trotz allem als nicht-invasiv gehandelt?
Vielen Dank vorab und beste Grüße,
Ewald Cerl
Sehr geehrter Herr Cerl,
danke für die spannende Frage!
Die „Invasivität“ vererbt sich nicht auf das jeweils andere Produkt.
Die Klassifizierungsregeln wollen ja sicherstellen, dass die speziellen Risiken bei der Klassifizierung berücksichtigt werden, die durch die Invasivität entstehen. Ein Produkt, das nicht invasiv ist, wird nicht dadurch invasiv, dass es mit einem invasiven Produkt gemeinsam verwendet werden muss. Das gilt auch in „umgekehrter Richtung“.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
gelten die in diesem Beitrag beschriebenen Anforderungen auch für IVDs? In der IVDD finde ich keinen entsprechenden Artikel dafür.
Wie ist vorzugehen, wenn IVDs kombiniert werden sollen?
Ein Kunde von uns will eines unserer CE gekennzeichneten IVDs in einen Kit mit anderen IVDs abpacken (die nicht von uns stammen) und zusätzlich den Intended Use ändern. Da der Intended Use geändert wird, wird demnach unser Kunde zum Hersteller des Kits. Da auf unserem Produkt im Kit jedoch weiterhin wir als Hersteller angeführt sind, haben wir bedenken, dass wir mit ungerechtfertigten Reklamationen konfrontiert werden könnten. Wie können wir das vermeiden bzw. wie können wir sicherstellen, dass wir für das Produkt im Kit keine Verantwortung übernehmen müssen?
Vielen Dank im Voraus für Ihre Hilfe!
Beste Grüße,
Maximilian
Sehr geehrter Herr Maximilian,
danke für die Nachfrage. Dazu zwei Antworten:
Danke für Ihr Nachfragen. Das ist wichtig, die Gemeinsamkeiten und Unterschiede zwischen MDR/IVDR zu beachten.
Viele Grüße, Christian Johner
Sehr geehrter Herr Johner,
mir ist die Zulässigkeit bzw. die Verantwortlichkeit für die Sicherheit von Kombinationen von Medizinprodukten noch nicht ganz klar. Ein Beispiel: Ein Hersteller möchte ein Medizinprodukt (Sensoren zur Überwachung) auf den Markt bringen, das in ein zweites Medizinprodukt eines anderen Herstellers eingebaut werden soll. Der Hersteller des zweiten Medizinprodukts hat allerdings nicht vorgesehen, dass das Medizinprodukt mit den Sensoren nachgerüstet wird.
Die Sensoren erweitern die Funktionalität des zweiten Medizinprodukts. Außerdem wird durch den Einbau zum Beispiel die Höhe des zweiten Medizinprodukts verändert. Beide Medizinprodukte sind mit CE gekennzeichnet. Wird durch diesen Zusammenbau ein neues Produkt erschaffen und muss anschließend neu Konformitätsbewertet werden? Das ist für den Hersteller der Sensoren, dem die technische Dokumentation des zweiten Medizinprodukts nicht vorliegt, doch gar nicht möglich. Muss der Hersteller der Sensoren für jede mögliche Kombination mit anderen Medizinprodukten eine neue Konformitätsbewertung durchlaufen oder reicht die Aussage, dass die Sensoren mit diesen Produkten kompatibel sind. Wie muss diese Kompatibilität nachgewiesen werden?
Wenn der Hersteller die die Sensoren auf den Markt bringt und ein Betreiber die beiden Medizinprodukte zusammenbaut, ist dieser dann allein für die Sicherheit verantwortlich?
Viele Grüße
Lasse Osterheider
Sehr geehrter Herr Osterheider,
danke für Ihre umfassende und spannende Frage.
Ich kenne die Details nicht, und gehe daher davon aus, dass die Sensoren tatsächlich Medizinprodukte sind. Oft sind das nur Komponenten oder Zubehör.
Wenn jemand, wie Sie schreiben, die Sensoren in ein Medizinprodukt einbaut, dann ist das ein neues Produkt und weder ein System noch eine Behandlungseinheit gemäß MDR. D.h. es ist eine neue Konformitätsbewertung notwendig.
Ich teile auch Ihre Einschätzung, dass das einem Hersteller, der nicht über die komplette Dokumentation beider „Komponenten“/Produkte verfügt, kaum möglich sein dürfte.
Wenn ein Betreiber diesen Zusammenbau macht, dann liegt eine Eigenherstellung vor. Aber genau diese wird durch die MDR sehr begrenzt. Sie ist nur statthaft, wenn es keine CE-gekennzeichnete Alternative auf dem Markt gibt.
Fazit: Wenn ein Szenario so abspielen würde, wie ich das verstehe, liegt mit einer nicht zu vernachlässigenden Wahrscheinlichkeit ein Rechtsbruch vor.
Beste Grüße, Christian Johner
Sehr geehrter Herr Johner,
seit Jahren begleitet uns folgende Frage zur Kopplung zweier zugelassener Medizinprodukte der Klasse 1:
Kann ein Patientenlifter des Herstellers A mit einem Hebetuch des Herstellers B kombiniert werden?
Sprich kann ein Patient im Hebetuch des Herstellers B sitzend mit dem Lifter des Herstellers A transferiert werden?
Vielen Dank im Voraus für Ihre Hilfe!
Beste Grüße,
Jacob Jansen
Sehr geehrter Herr Jansen,
es sollte grundsätzlich sichergestellt sein, dass die Kombination der zwei Produkte zu keinem Patientenrisiko führt und die Anwendung somit als „Sicher“ angesehen werden kann, unabhängig davon wer (Hersteller/Anwender) die Kombination durchführt. Sollte der Hersteller die Kombination in Verkehr bringen, muss die Kombination der Produkte entsprechend geprüft und dokumentiert werden.
Herzliche Grüße
Sehr geehrter Prof. Johner,
wie ist es wenn wir ein Produkt der Klasse II a (Bodyplethysmographen) mit einem BGA Gerät kombinieren?
Müssen wir die Risikobewertung durchführen? Oder können wir einfach sagen, dass wir keine Konfomität mehr erstellen können? Somit würde die Risokobewertung an das Haus übergeben.
Vielen Dank für die unterstützung!
Mit freundlichen Grüßen
Fuchs
Liebe/r Herr oder Frau Fuchs,
mein Kollege Mario Klessascheck hat mir dazu die folgende Antwort gegeben:
vielen Dank für Ihre Frage. Ich bin nicht sicher in welche Richtung Ihre Frage zielt. Sie sagen „keine Konfomität mehr erstellen können“. Ich vermute einen zeitlichen Aspekt. Geht es um die Übergangsfristen für altrechtliche Produkte? Oder geht es nur um das korrekte Vorgehen bzgl. der Verbindung? Es ist mir auch nicht klar, ob Sie als Betreiber oder Hersteller fragen. Ich vermute als Hersteller.
Wenn Sie Ihr Produkt mit einem BGA verbinden und diese Kombination in Verkehr bringen, liegt die Verantwortung vollständig bei Ihnen. Die Kombination darf für keines der beiden Produkte zu einem neuen Risiko führen, andernfalls hätten Sie ein neues Produkt gebaut. Wenn Ihr Produkt hingegen eine Schnittstelle hat, die dafür vorgesehen ist, ein BGA anzuschließen, dann haben Sie das typischerweise schon vor der Inverkehrbringung geprüft. Fragen Sie gern noch einmal nach, wenn meine Antwort nicht hilfreich war.
Liebe Grüße, MK