Die klinische Bewertung ist kein Dokument, sondern ein Prozess über den kompletten Produktlebenszyklus. Damit überprüfen die Hersteller Sicherheit, Leistungsfähigkeit und Wirksamkeit ihrer Produkte. Bei diesem Prozess entstehen Dokumente wie
- der Plan für die klinische Bewertung (Clinical Evaluation Plan) und
- der klinische Bewertungsbericht (Clinical Evaluation Report).
Inhalt
Diese Seite verschafft Ihnen einen schnellen Einstieg in das Thema und verlinkt relevante Fachartikel sowie weitere Hilfestellungen:
- Grundlagen
- Regulatorische Anforderungen
- Weitere Fachartikel
- Unterstützung bei der klinischen Bewertung
1. Klinische Bewertung: Die Grundlagen
a) Definition und Zielsetzung der klinischen Bewertung
Die MDR definiert den Begriff wie folgt:
„klinische Bewertung“ bezeichnet einen systematischen und geplanten Prozess zur kontinuierlichen Generierung, Sammlung, Analyse und Bewertung der klinischen Daten zu einem Produkt, mit dem Sicherheit und Leistung, einschließlich des klinischen Nutzens, des Produkts bei vom Hersteller vorgesehener Verwendung überprüft wird.
MDR Artikel 2, Satz 44
Die MDR legt damit auch die Zielsetzung fest: die Sicherheit, Leistungsfähigkeit und den klinischen Nutzen zu überprüfen. Daher ist die klinische Bewertung eine Voraussetzung für die Inverkehrbringung von Medizinprodukten.
Die klinische Bewertung muss anhand klinischer Daten erfolgen, um die notwendige klinische Evidenz zu erreichen. Manchmal wird in diesem Kontext auch von klinischer Validierung gesprochen.
Hinweise
Bei Medizinprodukten und In-vitro-Diagnostika IVD sind die Zweckbestimmung und die Medical Claims die Basis für die klinische Bewertung.
Bei IVD gibt es keine klinische Bewertung, sondern eine klinische Leistungsbewertung.
b) Die klinische Bewertung im Produktlebenszyklus
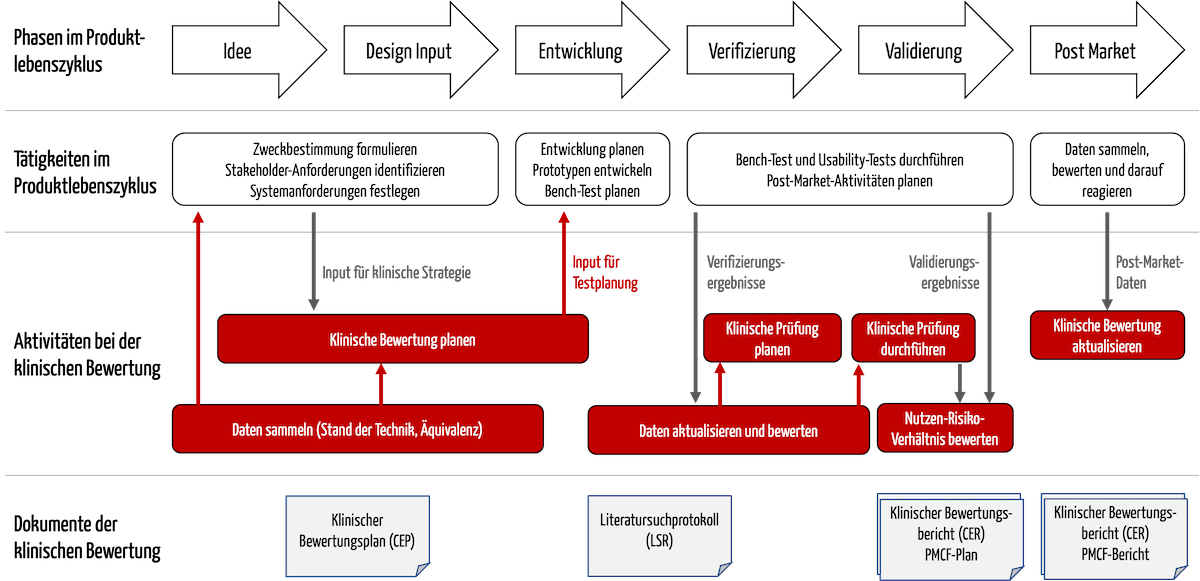
Die Aktivitäten bei der klinischen Bewertung sind eng mit weiteren Tätigkeiten im Produktlebenszyklus verzahnt, beispielsweise mit der Entwicklung (s. Abb. 1).
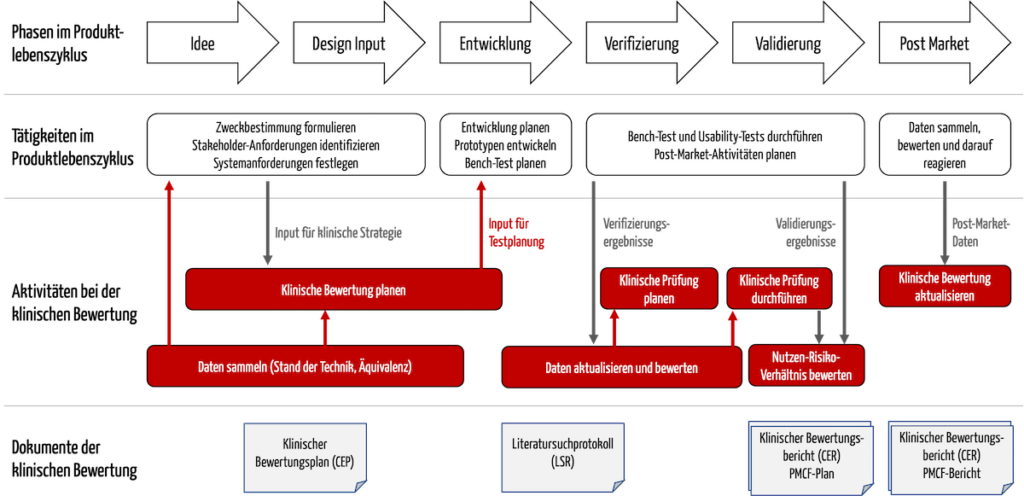
Abb. 1: Die Aktivitäten der klinischen Bewertungen interagieren mit der Entwicklung und der Post-Market Surveillance.
Tipp
Binden Sie bei der klinischen Bewertung nicht nur die Clinical Affairs Manager bzw. die Abteilung „Clinical Affairs“ ein, sondern fordern Sie Tätigkeiten auch von anderen Kompetenzen ein wie der Entwicklung, dem Produktmanagement sowie von Ärztinnen und Ärzten. Dazu ist auch die Rolle des Medical Writers einzubinden.
c) Vorteile einer professionellen klinischen Bewertung
Hersteller profitieren von einer professionellen und gesetzeskonformen klinischen Bewertung:
- Ärger bei Review, Audits und Zulassungen vermeiden
Die klinische Bewertungsakte steht im Zentrum fast aller Prüfungen. Abweichungen sind eher die Regel als die Ausnahme, können aber vermieden werden.
- Entwicklungsrisiken minimieren
Bei der klinischen Bewertung muss der Hersteller zu Beginn der Entwicklung den Stand der Technik recherchieren, auch hinsichtlich der Konkurrenz und der Leistungsfähigkeit der Produkte. Das wirkt sich u. a. darauf aus, ob klinische Prüfungen notwendig sind, die das Entwicklungsprojekt oft um Jahre verzögern. Regelmäßig scheitern Entwicklungen, weil diese Rahmenparameter zu spät erkannt werden.
- Produkte schneller in den Markt bringen
Eine rasche Entwicklung und eine reibungsfreie Zulassung führen zu einer schnellen Vermarktung und damit früh zu Umsätzen.
- Patientensicherheit verbessern
Die klinische Bewertung prüft definitionsgemäß, ob die Qualität der Produkte gegeben ist, sprich deren Sicherheit, Leistungsfähigkeit und klinischer Nutzen.
- Imageschäden und Kosten für Rückrufe vermeiden
Produkte mit einer hohen Qualität sind dem Image dienlich und lassen die Kosten für Produktrückrufe sinken.
- Marketingkommunikation zielgerichtet gestalten
Die klinische Bewertung stellt sicher, dass die Zweckbestimmung und der bestimmungsgemäße Gebrauch sehr präzise formuliert sind. Das bildet die Basis und den Rahmen für eine kohärente und gesetzeskonforme Marketingkommunikation.
2. Regulatorische Anforderungen an die klinische Bewertung
a) MEDDEV 2.7/1 revision 4
Bereits die Medizinprodukterichtlinie MDD forderte eine klinische Bewertung, allerdings eher unkonkret. Daher gibt es die MEDDEV-Leitlinie 2.7/1 (seit 2016 in der Revision 4), die genauere Handlungsanweisungen zur Durchführung der klinischen Bewertung gibt.
Die Leitlinie ist zwar nicht rechtlich bindend, aber bisher Goldstandard und Referenz für Inhalt und Aufbau von klinischen Bewertungen. Auditoren setzen die Einhaltung weiterhin meist voraus.
Auch die MDCG-Leitlinien verweisen auf die MEDDEV 2.7/1 Rev. 4, die damit nichts an Relevanz verliert.
b) MDR
Die MDR widmet der klinischen Bewertung den Artikel 61 sowie den Anhang XIV (Part A). Beide machen viele Anforderungen der MEDDEV 2.7.1, Rev. 4 rechtlich bindend.
Die MDR fordert als Teil der klinischen Bewertung einen Post-Market Clinical Follow-up (PMCF) sowie einen „Kurzbericht über Sicherheit und klinische Leistung“ (SSCP).
c) MDCG-Leitlinien
Zusätzlich hat die Medical Device Coordination Group (MDCG) mehrere Leitlinien zur klinischen Bewertung veröffentlicht. Tabelle 1 gibt eine Übersicht.
d) Zusammenfassung
| Regularie/Leitlinie |
Art / Relevanz / Wie hilft sie Ihnen? |
Prio |
| MDR (2017/745) |
Definiert die gesetzlichen Anforderungen, die jeder erfüllen muss. Sie gibt erste Instruktionen, wie die klinische Bewertung durchzuführen ist. |
1 |
| MEDDEV 2.7/1 rev. 4 |
Gibt wie ein „Kochbuch“ konkrete Anweisungen, wie der Prozess der klinischen Bewertung zu strukturieren und durchzuführen ist |
2-3 |
| MDCG 2020-1 |
Liefert eine detaillierte Interpretation der MDR bezüglich der klinischen Bewertung von Software als Medizinprodukt. |
2 |
| MDCG 2020-5 |
Enthält eine detaillierte Interpretation der MDR bezüglich der klinischen Bewertung auf der Grundlage der Äquivalenz zu anderen Produkten |
2 |
| MDCG 2020-6 |
Gibt eine detaillierte Interpretation der MDR bezüglich der klinischen Bewertung von Bestandsprodukten |
2 |
| MDCG 2020-13 |
Ist die „Checkliste“ (das CEAR-Template), das Auditoren benutzen, um die klinische Bewertung zu beurteilen
(CEAR = Clinical Evaluation Assessment Report) |
2 |
| MDCG 2024-10 |
Gibt Hilfestellung bei der klinischen Bewertung von Orphan Medical Devices |
2 |
| MDCG 2020-13 |
Ist die „Checkliste“ (das CEAR-Template), das Auditoren benutzen, um die klinische Bewertung zu beurteilen
(CEAR = Clinical Evaluation Assessment Report) |
2 |
| IMDRF MDCE WG/N56 |
Ein „Quick Guide“ mit konkreten Anweisungen, kürzer als die MEDDEV und kein Goldstandard, aber mit hilfreichen Tipps zur Literaturbeurteilung |
4 |
Tabelle 1: Die relevanten Regularien und Leitlinien zur klinischen Bewertung
4. Weitere Fachartikel
a) Fachartikel zu einzelnen Tätigkeiten
Die folgenden Artikel geben Hilfestellungen bei den aufgeführten Tätigkeiten:
b) Fachartikel zu klinischen Prüfungen
Verfügt ein Hersteller nicht über ausreichende klinische Daten, muss er eine klinische Prüfung durchführen.
c) Fachartikel zu regulatorischen Dokumenten und weiteren Themen
5. Unterstützung bei der klinischen Bewertung
Haben Sie noch Fragen zur klinischen Bewertung? Dann nutzen Sie das kostenfreie Micro-Consulting.
Die Expertinnen und Experten des Johner Instituts sind auf klinische Bewertungen spezialisiert und können
- Sie bei der klinischen Strategie unterstützen,
- Ihnen durch einen Quick-Check Klarheit über den Stand der eigenen klinischen Bewertung vermitteln,
- klinische Bewertungen erstellen und prüfen,
- mit Benannten Stellen Abweichungsberichte diskutieren und
- klinische Prüfungen planen und Fallzahlberechnungen durchführen.
Nehmen Sie Kontakt auf, um zu klären, wie Sie am schnellsten zu einer präzisen und gesetzeskonformen klinischen Bewertung kommen.
Kontakt aufnehmen
FAQ zur klinischen Bewertung
Benötigen Medizinprodukte der Klasse I eine klinische Bewertung?
Klinische Bewertung für Medizinprodukte der Klasse I
Alle Medizinprodukte müssen die grundlegenden Anforderungen erfüllen, die MDD und MDR fordern. Eine zentrale Rolle spielt in beiden Regularien die Forderung nach einer klinischen Bewertung (siehe auch MDR, Artikel 5, Abschnitt (3)).
Allerdings wird bei Klasse-I-Produkten wird die klinische Bewertung nicht von einer Benannten Stelle überprüft, auch nicht die gesamte technische Dokumentation. Jedoch kann die zuständige Regierungsbehörde jederzeit danach fragen.
Erlaubt die MDR den Verzicht auf klinische Daten?
Verzicht auf klinische Daten
Die MDR erlaubt lediglich unter ganz bestimmten Voraussetzungen, in der klinischen Bewertung auf klinische Daten zu verzichten: Die MDR erlaubt diese Ausnahme im Artikel 61, Satz (10), wenn der Nachweis mittels klinischer Daten als ungeeignet oder nicht angemessen erachtet wird („is not deemed appropriate“). Dieser Verzicht auf klinische Daten muss nachvollziehbar begründet werden
So können z.B. Hilfsmittel (z.B. Mundspatel) oder Werkzeuge (z.B. Skalpell, Zahnarztbohrer) oft ohne produktspezifische klinische Daten bewertet werden, da eine solitäre klinische Prüfung nicht angemessen oder ethisch nicht vertretbar erscheint. Natürlich muss dies nachvollziehbar begründet werden.
Für die Begründung können Sie z.B. über allgemeine klinische Daten im State of the Art nachweisen, dass Ihr Gerät auf einer bewährten Technologie basiert, die dem anerkannten Stand der Technik entspricht und allgemein wenig Änderung erfährt. Idealerweise liegen Produktnormen vor, in denen Leistungsparameter angegeben werden, durch deren Einhaltung die Sicherheit gewährleistet werden kann. Der Nutzen kann z.B. über Leitlinien medizinischer Fachgesellschaften nachgewiesen werden. Die MDCG beschreibt diese Möglichkeit nur im Dokument MDCG 2020-6 für Bestandsprodukte (englisch: legacy devices). Wir denken aber, dass der Grundgedanke auch anwendbar ist, wenn Sie ein derartiges Produkt neu in Ihr Portfolio aufnehmen und erstmals zulassen.
Vorsicht!
Beachten Sie, dass ein Verzicht auf klinische Daten nicht bedeutet, dass Sie Ihr Produkt nicht schon vor der Inverkehrbringung einer Prüfung durch Anwender unterziehen müssen. In den meisten Fällen brauchen Sie summative Tests, eine sogenannte „Usability-Studie“, um die Gebrauchstauglichkeit zu validieren.
Ersparen äquivalente Produkte die klinische Prüfung?
Keine klinische Prüfung bei Äquivalenzprodukten
Im Prinzip ja. Aber die Anforderungen an die Äquivalenzkriterien sind mittlerweile so hoch, dass sie fast nur im Falle von Vorgängerprodukten aus dem eigenen Haus erfüllt werden können. Sie müssen nämlich sehr detailliertes Wissen über die anderen Produkte nachweisen.
Für Klasse-III-Produkte und implantierbare Produkte werden in der MDR sogar explizit Verträge zwischen den Herstellern gefordert, die vollen Einblick in die technische Dokumentation gewähren.
Aber auch bei eigenen Vorgängerprodukten müssen Sie sehr genau prüfen. Als äquivalent gilt ein Produkt nur, wenn Sie sowohl technische als auch biologische und klinische Äquivalenz nachweisen können. Eine Änderung am Material oder an einer Beschichtung führt somit schnell zum Verlust der biologischen Äquivalenz, eine Erweiterung der Indikationen zum Verlust der klinischen Äquivalenz etc.
Sie brauchen nicht für jedes neue Produkt automatisch eine Studie. Prüfen Sie, ob für Ihr Produkt klinische Daten überhaupt notwendig bzw. geeignet sind, um die Erfüllung der grundlegenden Anforderungen an Leistung und Sicherheit nachzuweisen.
Ist die klinische Bewertung ein Dokument?
Dokument oder Prozess
Die klinische Bewertung ist kein Dokument, sondern ein Prozess. Die MDCG schreibt hierzu im Dokument MDCG 2020-13:
„ … the clinical evaluation report (CER) and the related clinical evaluation that was conducted – a core requirement of the Medical Device Regulation (EU) 2017/745 (MDR).”
Neben dem Plan zur klinischen Bewertung, der schon in der MDR ganz klar gefordert wird, erweitert das Dokument MDCG 2020-13 die klinische Bewertungsakte noch um einen Plan und einen Bericht zur Literaturrecherche.
Lesen Sie hier mehr dazu, wie diese beiden Dokumente entstehen.
Wann wird die klinische Bewertung geschrieben? Am Ende der Produktentwicklung?
Zeitpunkt der Erstellung
Nein! Der Prozess der klinischen Bewertung kann gar nicht früh genug begonnen werden. Denn sie ist nicht nur wichtig für die Planung, sondern auch eine wesentliche Voraussetzung für das Risikomanagement. Erst die klinische Bewertung rechtfertigt die in der Risikomanagementakte getroffenen Annahmen bezüglich des Nutzens und damit der Akzeptanz eines bestimmten Nutzen-Risiko-Verhältnisses. Die klinische Bewertung muss auch die Annahmen in der Risikomanagementakte bezüglich der klinischen Risiken bestätigen.
Allerdings ist der finale Bericht zur klinischen Bewertung eines der letzten Dokumente, die für die Zulassung eines Medizinprodukts fertig gestellt werden.
Wie spielt die klinische Bewertung mit anderen Dokumenten zusammen? Referenziert sie diese, wird selbst aber nicht referenziert?
Zusammenspiel mit anderen Dokumenten
Die klinische Bewertung ist kein Quelldokument für Produktinformationen, sondern eine Zusammenfassung und Bewertung. In den Dokumenten der klinischen Bewertung wird in der Tat viel auf andere Dokumente verwiesen.
Informationen wie die Zweckbestimmung müssen in allen Dokumenten den gleichen Wortlaut haben. Sie sollten immer die Referenz auf das Quelldokument angeben. Die Berichte, denen Sie die zu bewertenden Daten entnehmen, werden Sie meist zusammenfassen und ebenfalls auf die Quellen referenzieren.
Tipp
Der klinische Bewertungsbericht sollte ein eigenständiges, für sich lesbares Dokument sein. Welche Informationen die benannte Stelle für den Review Ihrer klinischen Bewertung auf jeden Fall benötigt, sehen Sie im Dokument MDCG 2020-13, dem Template für den „Clinical Evaluation Report“ (CER), den der Auditor bzw. Reviewer anfertigen muss.