
Betriebssystem konform mit IEC 62304 und FDA?
Müssen Medizinproduktehersteller bei der Auswahl des Betriebssystems darauf achten, dass das Betriebssystem IEC- 62304-konform ist? Was sagt die FDA? Dieser Artikel …

Zur medizinischen Software zählt alle Software, die für das Gesundheitswesen eingesetzt wird, insbesondere Software für Medizinprodukte bzw. Medizingeräte (Embedded Software) und Software, die selbst ein Medizinprodukt ist (Standalone-Software).
Die IEC/CD1 82304-1 (Health Software – Part 1: General requirements for product safety) unterscheidet folgende Begriffe:
Damit wird klar, dass medizinische Software ein Medizinprodukt sein kann, aber nicht muss.
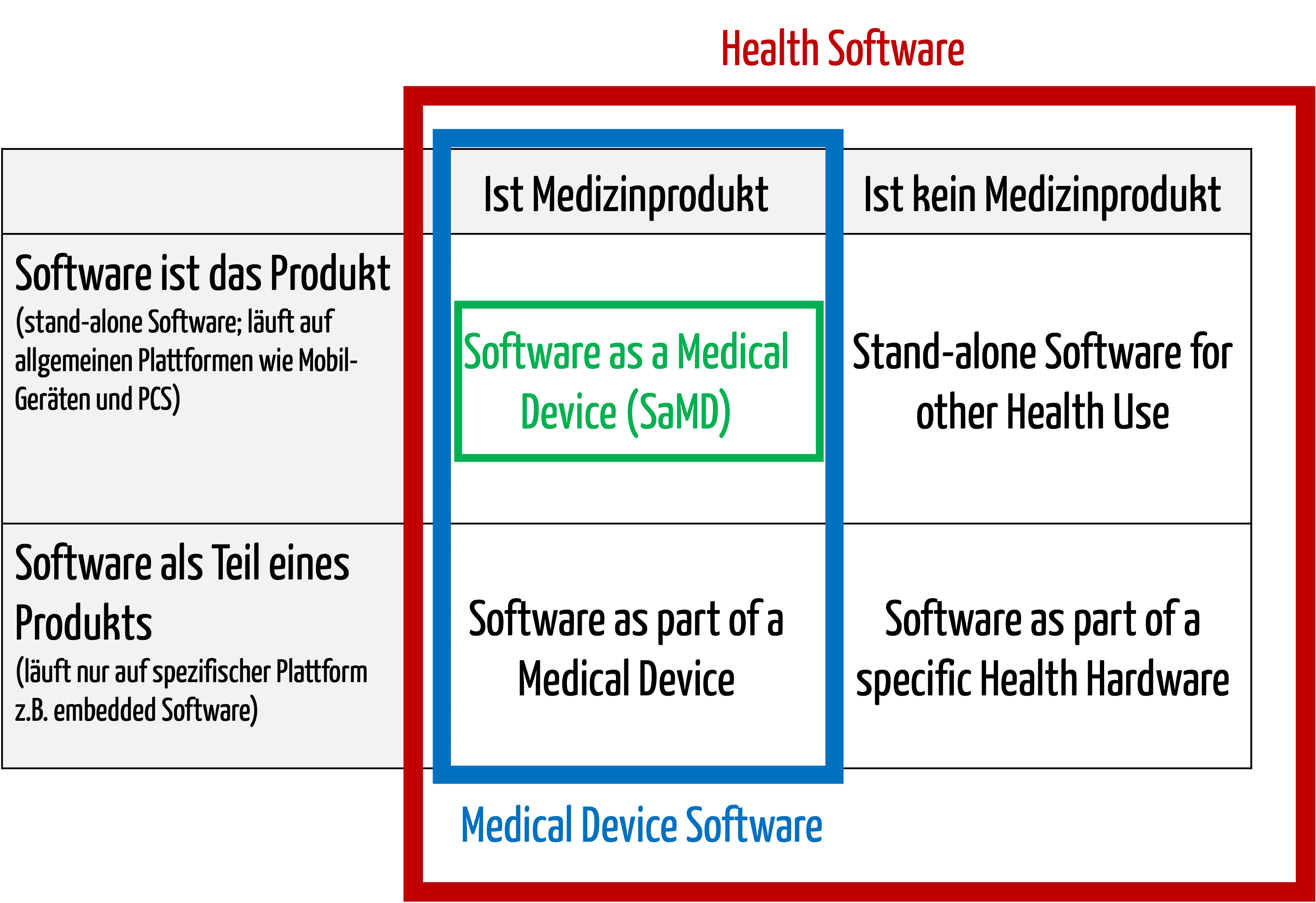
Abb. 1: Medizinische Software umfasst auch Medical Device Software und Software as a Medical Device (zum Vergrößern klicken).
Es stellt sich oft die Frage, wann medizinische Software für die Medizintechnik der Definition des Begriffs Medizinprodukt entspricht. Eine weiterführende Diskussion dazu finden Sie im Artikel zur Klassifizierung von Software als Medizinprodukt sowie im Artikel zur Qualifizierung und Klassifizierung von IVD-Software.
Software, die ein Medizinprodukt oder ein Teil dessen ist, muss die regulatorischen Anforderungen erfüllen:
Lesen Sie hier mehr zum Thema gesetzeskonforme Software-Entwicklung und IEC 62304. Beachten Sie auch den Podcast zum Thema medizinische Software. Beachten Sie auch den Artikel zu den klinischen Informationssystemen z.B. zu den Patientendaten-Managementsystemen (PDMS).
Nutzen Sie die Unterstützung des Johner Instituts:
Melden Sie sich gleich, damit wir die nächsten Schritte besprechen können. So stellen Sie sicher, dass die „Zulassung“ sicher gelingt und Ihre Software bzw. Ihre Produkte schnell in den Markt kommen.
Müssen Medizinproduktehersteller bei der Auswahl des Betriebssystems darauf achten, dass das Betriebssystem IEC- 62304-konform ist? Was sagt die FDA? Dieser Artikel …

Konfigurationsmanagement ist weit mehr als nur der Einsatz von Versionsverwaltungswerkzeugen wie git oder svn. Dies wird bei einem Blick in die IEC 62304 und die Guidance-Dokumente der FDA sofort klar. Lesen Sie in diesem Artikel,
Details
Die zyklomatische Komplexität ist eine Metrik im Software-Engineering, welche die Komplexität und damit die Fehlerträchtigkeit und Wartbarkeit von Code zu bestimmen hilft. Die zyklomatische Komplexität wird auch als Mc Cabe Maß bezeichnet.

Elektronische Unterschriften und digitale Signaturen dürfen als gleichwertig zu handgeschriebenen Unterschriften („wet ink“) betrachtet werden. Welche Voraussetzungen dafür erfüllt sein müssen, hängt vom Grad der zu erreichenden Verbindlichkeit und damit vom Dokument ab, das unterschrieben werden soll. Dieser Artikel klärt,
DetailsDie ISO 27001 und die Informationssicherheitsmanagementsysteme (ISMS) werden bei Medizinprodukteherstellern immer häufiger zum Thema. Die Regularien geben dazu Anlass. Dazu zählt u. a. die Digitale-Gesundheitsanwendungen-Verordnung (DiGAV), die die ISO 27001 in den Fokus vieler Medizinproduktehersteller gerückt hat. Hersteller müssen die regulatorischen Anforderungen erfüllen, um Ärger mit Behörden und Benannten Stellen zu vermeiden und um Patienten…
Details
PDMS steht für Patientendatenmanagementsystem. Diese klinischen Informationssysteme finden sich typischerweise in Krankenhäusern, v. a. in den Abteilungen, die Patienten intensivmedizinisch behandeln. Durch die Förderungen des Krankenhaus-Zukunftsgesetzes (KHZG) erleben die PMDS einen neuen Boom. Dieser Artikel
DetailsDie Telematikinfrastruktur (TI) ist eine Plattform bzw. ein Netzwerk, über das in Deutschland Gesundheitsdaten sicher ausgetauscht werden und auf der Gesundheitsanwendungen wie die elektronische Patientenakte und das e-Rezept betrieben werden sollen. Dieser Beitrag hilft Herstellern medizinischer Software zu verstehen, wann sie welche gesetzlichen und technischen Anforderungen im Kontext dieser Telematikinfrastruktur erfüllen müssen.
DetailsWie unterscheiden sich Verifizierung und Validierung und wie sind diese Begriffe definiert? Selbst Normen und Gesetze verwenden die Begriffe falsch oder missverständlich. Dieser Artikel
DetailsMedizinprodukte- und IVD-Hersteller verwenden zunehmend Cloud-Dienste: Erfahren Sie, welche Möglichkeiten Hersteller haben, um Cloud-Dienste wie Medical Clouds zu nutzen und dennoch die regulatorischen Anforderungen an z. B. den Datenschutz zu erfüllen.
DetailsGesetze fordern das Risikomanagement im Krankenhaus, vor allem, um die Patientensicherheit zu verbessern. Dennoch tun sich viele Krankenhäuser damit schwer. Dieser Artikel stellt die wichtigsten regulatorischen Anforderungen vor und gibt Tipps zur Umsetzung.
DetailsWir nutzen Cookies auf unserer Website. Einige von ihnen sind essenziell, während andere uns helfen, diese Website und Ihre Erfahrung zu verbessern. Wenn Sie unter 16 Jahre alt sind und Ihre Zustimmung zu freiwilligen Diensten geben möchten, müssen Sie Ihre Erziehungsberechtigten um Erlaubnis bitten. Wir verwenden Cookies und andere Technologien auf unserer Website. Einige von ihnen sind essenziell, während andere uns helfen, diese Website und Ihre Erfahrung zu verbessern. Personenbezogene Daten können verarbeitet werden (z. B. IP-Adressen), z. B. für personalisierte Anzeigen und Inhalte oder Anzeigen- und Inhaltsmessung. Weitere Informationen über die Verwendung Ihrer Daten finden Sie in unserer Datenschutzerklärung. Sie können Ihre Auswahl jederzeit unter Einstellungen widerrufen oder anpassen.
Wenn Sie unter 16 Jahre alt sind und Ihre Zustimmung zu freiwilligen Diensten geben möchten, müssen Sie Ihre Erziehungsberechtigten um Erlaubnis bitten. Wir verwenden Cookies und andere Technologien auf unserer Website. Einige von ihnen sind essenziell, während andere uns helfen, diese Website und Ihre Erfahrung zu verbessern. Personenbezogene Daten können verarbeitet werden (z. B. IP-Adressen), z. B. für personalisierte Anzeigen und Inhalte oder Anzeigen- und Inhaltsmessung. Weitere Informationen über die Verwendung Ihrer Daten finden Sie in unserer Datenschutzerklärung. Hier finden Sie eine Übersicht über alle verwendeten Cookies. Sie können Ihre Einwilligung zu ganzen Kategorien geben oder sich weitere Informationen anzeigen lassen und so nur bestimmte Cookies auswählen.