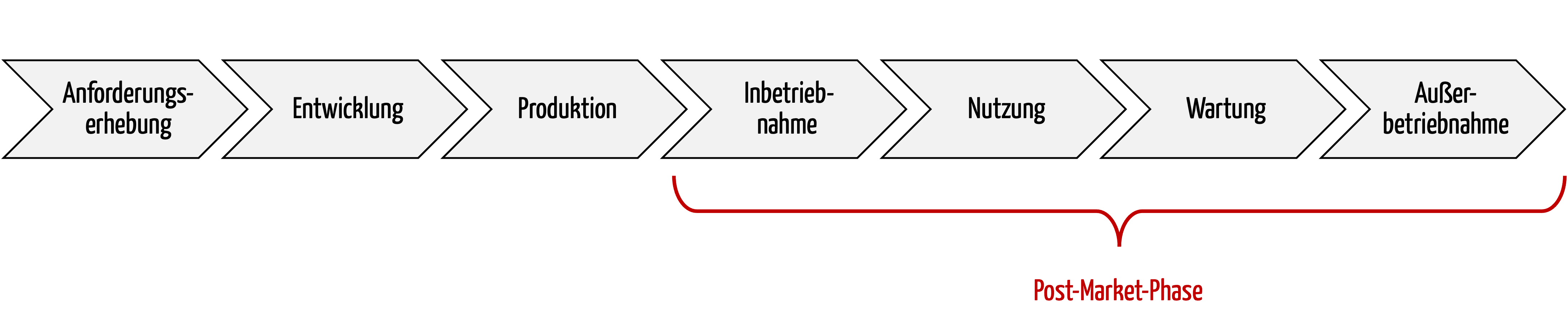
Die Post-Market-Phase ist die Phase nach der Inverkehrbringung von Medizinprodukten. Die ISO 14971 spricht auch von der der Entwicklung und Produktion nachgelagerten Phase.
Inhalt
Diese Seite verschafft Ihnen einen schnellen Überblick über die Fachartikel zur Post-Market Phase:
- Lebenszyklus von Medizinprodukten
- Regulatorische Anforderungen
- Aktivtäten in der Post-Market-Phase
- Verantwortliche Rollen
- Zusammenspiel der Post-Market Phase mit dem Risikomanagement
- Unterstützung
1. Lebenszyklus von Medizinprodukten
Die Post-Market-Phase bzw. nachgelagerte Phasen umfasst alle Aktivitäten nach der Produktion bis zur Stilllegung / Außerbetriebnahme der Produkte, spätestens am Ende der Lebensdauer der Produkte. Zu diesen Aktivitäten zählen auch die Nutzung und die Wartung.
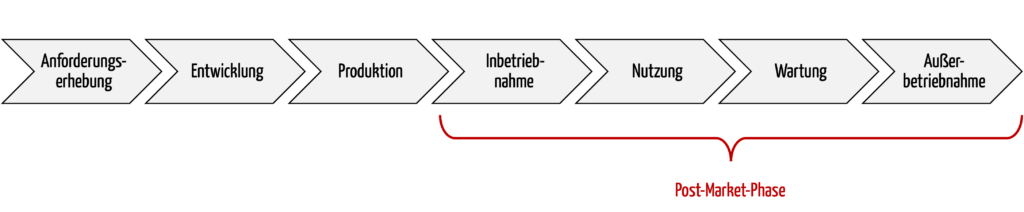
Abb. 1: Ein wichtiger Teil des Produktlebenszyklus ist die Post-Market-Phase.
2. Regulatorische Anforderungen an die Post-Market-Phase
Die internationalen regulatorischen Anforderungen an diese Phase steigen. Davon zeugen die EU-Verordnungen MDR und IVDR. Dazu kommen nationale Vorschriften wie er 21 CFR part 822 zur Post-Market Surveillance und nationale Gesetze und Verordnungen zur Vigilanz wie z.B. die MPAIMV.
Zudem gibt es Leitlinien z.B. die MDCG 2022-21, die MDCG 2020-6 und Gesetze, die für alle Produkt gelten wie das Produkthaftungsgesetz, das ebenfalls Post-Market-Aktivitäten wie die Überwachung vorsieht.
3. Aktivitäten in der Post-Market-Phase
a) Post-Market Surveillance
Die Regularien verpflichten die Hersteller beispielweise zur Post-Market Surveillance. Dafür müssen die Hersteller einen Post-Market-Surveillance-Plan erstellen und gemäß diesem die Post-Market-Surveillance-Daten sammeln und auswerten. Besonders schwer tun sich die Hersteller dabei mit der Trendanalyse. Die Ergebnisse münden dann in Berichten wie dem Periodic Safety Update Report (PSUR).
Abhängig von der Klasse der Produkte müssen die Berichte auch in der EUDAMED publiziert werden.
b) Post-Market Clinical Follow-up
Ein Teil der Post-Market Surveillance ist der Post-Market Clinical Follow-up (PMCF). Dabei werden klinische Daten gesammelt, um die klinische Bewertung aktuell zu halten. Sind nicht ausreichend Daten vorhanden, so können PMCF-Studien notwendig werden.
c) SSCP
Die MDR fordert als Teil der klinischen Bewertung einen „Kurzbericht über Sicherheit und klinische Leistung“ (SSCP). Bei der IVDR heißt dieser SCP.
d) Vigilanz
Falls in der Post-Market-Phase schwerwiegende Vorkommnisse oder Trends, die zu einem nicht-akzeptablen Risiko führen, beobachtet werden, sind die Hersteller zu Behördenmeldungen (Vigilanz) und zu Korrekturmaßnahmen verpflichtet.
e) Sonstiges
Die Überwachung neuer Regularien (Regulatory Update) zählt ebenso zu den Post-Market-Aktivitäten wie das Monitoring von Schwachstellen der Off-the-Shelf-Software und die Beseitigung dieser Schwachstellen mit Security-Patches.
4. Verantwortliche Rollen
Für diese Tätigkeiten sind nicht nur die Hersteller verantwortlich, beispielsweise deren Produktmanager, Qualitätsmanagementbeauftragten, Medizinprodukteberater und verantwortlichen Personen.
Auch die Lieferanten, Importeure und Händler haben Pflichten in dieser nachgelagerten Phase.
5. Zusammenspiel mit dem Risikomanagement
Das wesentliche Ziel all dieser Tätigkeiten besteht darin, die Sicherheit der Patienten zu gewährleisten, d.h. Risiken durch unsichere Produkte zu minimieren.
Daher hat die dritte Ausgabe der Risikomanagementnorm ISO 14971 die Anforderungen an die nachgelagerte Phase erhöht. Der zugehörige Technical Report ISO/TR 24971 beschreibt diese Anforderungen noch genauer.
Um das Nutzen-Risiko-Verhältnis abzuschätzen, müssen die Hersteller kontinuierlich den Stand der Technik verfolgen und berücksichtigen.
All dies nicht zu tun, zählt zu den häufigsten Fehlern beim Risikomanagement.
6. Unterstützung
Haben Sie noch Fragen zur Post-Market-Phase? Dann nutzen Sie das kostenfreie Micro-Consulting.
Falls Sie sich tiefer in das Thema einarbeiten wollen, dann bieten sich an:
- Das Seminar „Post-Market Surveillance“
- Die Videotrainings in der Johner Academy
Die Post-Market-Services nehmen Ihnen einen großen Teil der Arbeit ab: vom Überwachen der Regularien bis zur Post-Market Surveillance. Damit erreichen Sie:
- Regulatorische Sicherheit und Compliance
- Zuverlässige, schnelle und umfassende Überwachung und Reaktion
- Nachverfolgung der Maßnahmen
- Kostenreduktion durch Automatisierung
- Zeit für wertschöpfende Aufgaben (zur Kompensation des Fachkräftemangels)
Melden Sie sich, wenn Sie Unterstützung wünschen.